|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2011 | Volume
: 17
| Issue : 1 | Page : 29-32 |
| |
MCT oil-based diet reverses hypertrophic cardiomyopathy in a patient with very long chain acyl-coA dehydrogenase deficiency
Muhammad Ali Pervaiz1, Fran Kendal2, Madhuri Hegde1, Rani H Singh1
1 Department of Human Genetics, Emory University School of Medicine, 2165 North Decatur Road, Decatur, GA 30033, USA
2 Virtual Medical Practice, 5579 Chamblee Dunwoody Road, Suite 110, Atlanta, GA 30338-4128, USA
| Date of Web Publication | 18-Jun-2011 |
Correspondence Address:
Muhammad Ali Pervaiz
Department of Laboratory Medicine and Pathology, Mayo Clinic, 200 First Street SW, Rochester, MN 55905
USA
 Source of Support: None, Conflict of Interest: None  | 4 |
DOI: 10.4103/0971-6866.82190

 Abstract Abstract | | |
Very long chain acyl-CoA dehydrogenase (VLCAD) deficiency is one of the genetic defects of mitochondrial fatty acid beta-oxidation presenting in early infancy or childhood. If undiagnosed and untreated, VLCAD deficiency may be fatal, secondary to cardiac involvement. We assessed the effect of replacing part of the fat in the diet of a 2 -month-old male infant, who was diagnosed with VLCAD deficiency,with medium-chain triglyceride (MCT) oil and essential fats. The patient presented with vomiting, dehydration, and was found to have persistent elevation of liver function tests, hepatomegaly, pericardial and pleural effusion, right bundle branch block, and biventricular hypertrophy. Because of the cardiomyopathy, hepatomegaly, and an abnormal acylcarnitine profile and urine organic acids, he was suspected of having VLCAD deficiency. This was confirmed on acyl-coA dehydrogenase, very long chain (ACADVL) gene analysis. He was begun on an MCT oil-based formula with added essential fatty acids, uncooked cornstarch (around 1 year of age), and frequent feeds. By 7 months of age, cardiomyopathy had reversed and by 18 months of age, all cardiac medications were discontinued and hypotonia had improved such that physical therapy was no longer required. At 5 years of age, he is at the 50 th percentile for height and weight along with normal development. Pediatricians need to be aware about the basic pathophysiology of the disease and the rationale behind its treatment as more patients are being diagnosed because of expansion of newborn screen. The use of MCT oil as a medical intervention for treatment of VLCAD deficiency remains controversial mostly because of lack of clear phenotype-genotype correlations, secondary to the genetic heterogeneity of the mutations. Our case demonstrated the medical necessity of MCT oil-based nutritional intervention and the need for the further research for the development of specific guidelines to improve the care of these patients.
Keywords: Cardiomyopathy, carnitine, inborn error of metabolism, MCT oil, very long chain acyl-CoA dehydrogenase deficiency
How to cite this article:
Pervaiz MA, Kendal F, Hegde M, Singh RH. MCT oil-based diet reverses hypertrophic cardiomyopathy in a patient with very long chain acyl-coA dehydrogenase deficiency. Indian J Hum Genet 2011;17:29-32 |
How to cite this URL:
Pervaiz MA, Kendal F, Hegde M, Singh RH. MCT oil-based diet reverses hypertrophic cardiomyopathy in a patient with very long chain acyl-coA dehydrogenase deficiency. Indian J Hum Genet [serial online] 2011 [cited 2016 May 13];17:29-32. Available from: http://www.ijhg.com/text.asp?2011/17/1/29/82190 |
| Introduction | |  |
Very long chain acyl-CoA dehydrogenase (VLCAD) is the first enzyme in the beta oxidation of fatty acids once they have been transported inside the mitochondria; [1],[2] the enzyme lies on the inner mitochondrial membrane. During times of high energy requirements, long chain fatty acids are mobilized from adipose tissue and eventually metabolized by the heart and muscle. There are three phenotypes associated with this enzyme deficiency based on age of presentation and organ system involvement. [3] In this case report, we describe a patient with VLCAD deficiency diagnosed symptomatically after he was found to have hepatomegaly and cardiomyopathy.
| Case Report | | |
Our patient was a 2½-month-old male infant who presented to the hospital because of persistent emesis for 24 hours. He was also found to be hyponatremic, dehydrated, and hypotensive. The patient was admitted for rehydration and needed cardiac support with dopamine. He was also worked up for sepsis and, although one culture was positive, it was believed to be contamination. The remaining cultures were negative. Because his liver enzymes were persistently elevated, he underwent an abdominal ultrasound which showed hepatomegaly and pleural effusion. This led to a chest x-ray which revealed an enlarged cardiac shadow; hypertrophic cardiomyopathy with decreased ejection fraction and pericardial effusion were diagnosed by echocardiogram. An electrocardiogram showed right bundle branch block. There was no evidence of hypoglycemia during the hospital stay, but the patient was on dextrose drip from the time of admission which could have prevented this. His urine was positive for ketones. Unfortunately, no blood creatine phosphokinase (CPK) levels were checked at this time. His acylcarnitine profile [Table 1], done as part of a metabolic work up, was consistent with VLCAD deficiency.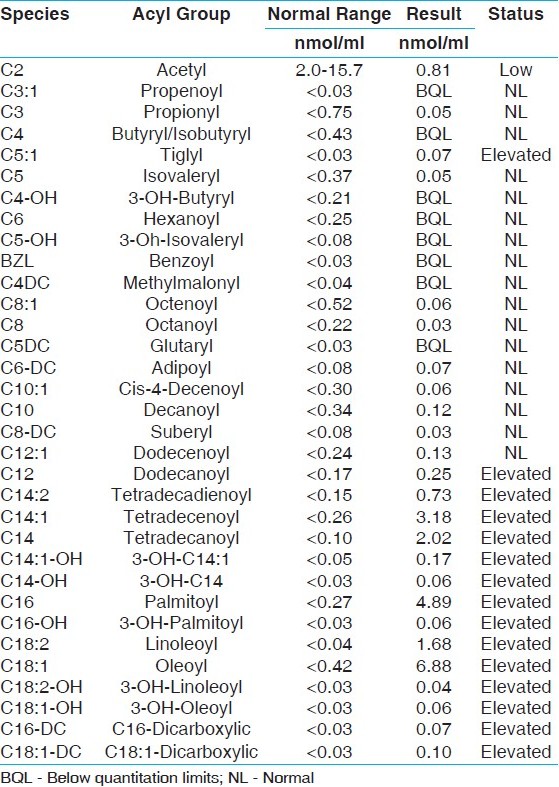 | Table 1: Acylcarnitine profi le showing marked elevation of long chain species, especially C14 complex, C16, C18:1 in a pattern consistent with VLCAD deficiency
Click here to view |
To treat this patient, the pericardial effusion was drained. As he became rehydrated, his cardiac pressors were stopped. He was started on a medium chain triglyceride (MCT) oil-based formula (Portagen) as part of his treatment. [4] Except for essential fatty acids, long-chain fatty acids were avoided. After two weeks of inpatient treatment, the patient was discharged on captopril, lasix, aldactone, potassium, riboflavin, and carnitine. He was continued on an MCT oil-based diet (total fat calories came 60% from MCT and 40% from long-chain triglycerides; these calories from fat represent 40% of his total energy requirement). A repeat echo done at 18 months of age showed complete resolution of the cardiomyopathy. The patient continued on an MCT oil-based diet and had a G-tube because of the taste of the formula. All his cardiac medications had been stopped, and he has regular cardiology follow-up. His creatinine phosphokinase levels continue to fluctuate with his activity level. His carnitine dose is adjusted periodically dependent on his plasma carnitine levels. We aim to keep it within normal ranges and stop it if it is higher than that.
Acyl-CoA dehydrogenase, long chain (ACADVL) [5] gene sequencing revealed an apparently homozygous c.1141_1143delGAG in-frame deletion in exon 11. Targeted comparative genomic hybridization (CGH) array for the ACADVL gene identified a deletion mutation encompassing exon 8 to exon 18 (partial), with approximate genomic breakpoints at nucleotide positions g.7,065,899 in intron 7 and g.7,068,084 in exon 18. Both of these mutations are expected to make a truncated protein. Subsequent analysis identified one copy of the c.1141_1143delGAG in-frame deletion in this individual's mother. The c.1141_1143delGAG mutation was not found in this individual's father, and we did not perform targeted CGH array analysis on the father, though we suspect that he might be a carrier of the partial deletion mutation encompassing exon 8 to exon 18.
| Discussion | | |
VLCAD deficiency can present in one of the following three forms: (1) severe, early-onset with cardiac and multiorgan failure VLCAD-C, [6],[7] (2) early childhood form with hypoketotic hypoglycemia and hepatomegaly VLCAD-H, and (3) the later-onset episodic myopathic form VLCAD-M. Clinically, our patient fits the first phenotype. He had a typical presentation consisting of hepatomegaly, cardiomyopathy, and cardiac arrhythmias. We did not record any hypoglycemic episodes during his hospital stay, though he was on a dextrose drip as part of his rehydration.
Treatment is essentially dietary modification, with avoidance of long-chain fatty acids and supplementation with medium chain triglycerides, so that the enzyme-deficient step can be bypassed. [8] Such treatment should reverse most symptoms, although during times of stress, like exercise, the MCT dosage may need to be raised to supply the extra energy. This helps prevent mobilization of long-chain fatty acids from the adipose tissue. Fasting needs to be avoided for similar reasons. [9] Close follow-up with a metabolic specialist and nutritionist is important to optimize dietary management. Acute rhabdomyolysis may occur with any form of VLCAD deficiency and must be treated aggressively because of the danger of renal failure. [10] Also, CPK along with CK-MB (creatinine phosphokinase, muscle and brain fractions) need to be monitored. Any siblings of the patient should be evaluated, as the symptoms may be subtle. [11]
In our clinic, an MCT oil-based diet is the main therapy for all patients with VLCAD deficiency. This treatment is based on our understanding of the pathophysiology of the condition. A recent DELPHI protocol recommends treatment based on VLCAD type and age. [12] There was no consensus on the treatment for asymptomatic VLCAD-C patients who are being breast fed and are less than 12 months of age. A European consensus report similarly could not come to a conclusion. [13] Patient with symptomatic VLCAD-C deficiency should have maximal MCT oil-based nutrition in both of these recommendations. The use of MCT oil as a medical intervention VLCAD deficiency remains controversial mostly because of lack of clear phenotype-genotype correlations, secondary to the genetic heterogeneity of the mutations. [14] Though genotype-phenotype correlation have been reported in the past, [15],[16] these are based on patients who were diagnosed symptomatically.
Because of the expanded newborn screen and its implementation in parts of the world, more pediatricians are going to be involved in the care of these patients, and this report will help them understand the basis of treatment for these patients. We feel that once we have genotype-phenotype information available on patients diagnosed through newborn screen, we will be able to improve the treatment further and provide personalized interventions. Based on such information, further clinical studies with a larger sample size will help determine optimal nutrition care for patients with VLCAD deficiency, as well as the role of MCT oil supplementation.
| References | | |
| 1. | Aoyama T, Uchida Y, Kelley RI, Marble M, Hofman K, Tonsgard JH, et al. A novel disease with deficiency of mitochondrial very-long-chain acyl-CoA dehydrogenase. Biochem Biophys Res Commun 1993;191:1369-72. 
|
| 2. | Aoyama T, Souri M, Ushikubo S, Kamijo T, Yamaguchi S, Kelley RI, et al. Purification of human very-long-chain acyl-coenzyme A dehydrogenase and characterization of its deficiency in seven patients. J Clin Investig 1993;95:2465-73.
|
| 3. | Vianey-Saban C, Divry P, Brivet M, Nada M, Zabot MT, Mathieu M, et al. Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: Clinical characteristics and diagnostic considerations in 30 patients. Clin Chim Acta 1998;269:43-62.
|
| 4. | Solis JO, Singh RH. Management of fatty acid oxidation disorders: A survey of current treatment strategies. J Am Diet Assoc 2002;102:1800-3.
|
| 5. | Andresen BS, Bross P, Vianey-Saban C, Divry P, Zabot MT, Roe CR, et al. Cloning and characterization of human very long-chain acyl- CoA dehydrogenase cDNA, chromosomal assignment of the gene, and identification in four patients of nine different mutations within the VLCAD gene. Hum Mol Genet 1996;5:461-72.
|
| 6. | Parini R, Menni F, Garavaglia B, Fesslova V, Melotti D, Massone ML, et al. Acute, severe cardiomyopathy as main symptom of late-onset very long-chain acyl-coenzyme A dehydrogenase deficiency. Eur J Pediatr 1998;157:992-5.
|
| 7. | Mathur A, Sims HF, Gopalakrishnan D, Gibson B, Rinaldo P, Vockley J, et al. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation 1999;99:1337-43.
|
| 8. | Cox GF, Souri M, Aoyama T, Rockenmacher S, Varvogli L, Rohr F, et al. Reversal of severe hypertrophic cardiomyopathy and excellent neuropsychologic outcome in very-long chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr 1998;133:247-53.
|
| 9. | Roe CR, Wiltse HE, Sweetman L, Alvarado LL. Death caused by preoperative fasting and sedation in a child with unrecognized very long-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr 2000;136:397-9.
|
| 10. | Laforêt P, Acquaviva-Bourdain C, Rigal O, Brivet M, Penisson-Besnier I, Chabrol B, et al. Diagnostic assessment and long-term follow-up of 13 patients with Very Long-Chain Acyl-Coenzyme A dehydrogenase (VLCAD) deficiency. Neuromuscul Disord 2009;19:324-9.
|
| 11. | Sluysmans T, Tuerlinckx D, Hubinont C, Verellen-Dumoulin C, Brivet M, Vianey-Saban C. Very long chain acyl-coenzyme A dehydrogenase deficiency in two siblings: Evolution after prenatal diagnosis and prompt management. J Pediatr 1997;131:444-6.
|
| 12. | Arnold GL, Van Hove J, Freedenberg D, Strauss A, Longo N, Burton B, et al. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab 2009;96:85-90.
|
| 13. | Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, et al. Treatment recommendations in long-chain fatty acid oxidation defects: Consensus from a workshop. J Inherit Metab Dis 2009;32:498-505.
|
| 14. | Pons R, Cavadini P, Baratta S, Invernizzi F, Lamantea E, Garavaglia B, et al. Clinical and molecular heterogeneity in very-long-chain acyl-coenzyme A dehydrogenase deficiency. Pediatr Neurol 2000;22:98-105.
|
| 15. | Andresen BS, Olpin S, Poorthuis BJ, Scholte HR, Vianey-Saban C, Wanders R, et al. Clear correlation of genotype with disease phenotype in very long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet 1999;64:479-94.
|
| 16. | Gobin-Limballe S, McAndrew RP, Djouadi F, Kim JJ, Bastin J. Compared effects of missense mutations in Very-Long-Chain Acyl-CoA Dehydrogenase deficiency: Combined analysis by structural, functional and pharmacological approaches. Biochim Biophys Acta 2010;1802:478-84.
|
[Table 1]
| This article has been cited by | | 1 |
Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD-/-) mice |
|
| Sara Tucci,Ulrich Flögel,Sven Hermann,Marga Sturm,Michael Schäfers,Ute Spiekerkoetter | | Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2014; 1842(5): 677 | | [Pubmed] | [DOI] | | | 2 |
Evaluation of cardiac manifestations in pediatric liver transplant candidates |
|
| Nitin Madan,Ronen Arnon,Rica Arnon | | Pediatric Transplantation. 2012; 16(4): 318 | | [Pubmed] | [DOI] | | | 3 |
Evaluation of cardiac manifestations in pediatric liver transplant candidates |
|
| Madan, N. and Arnon, R. and Arnon, R. | | Pediatric Transplantation. 2012; 16(4): 318-328 | | [Pubmed] | |
|
|
|
|