|
 
 |
|
ORIGINAL ARTICLE |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 4 | Page : 449-453 |
| |
Mutational identification of fibroblast growth factor receptor 1 and fibroblast growth factor receptor 2 genes in craniosynostosis in Indian population
Rajeev Kumar Pandey, Minu Bajpai, Abid Ali, Sukanya Gayan, Amit Singh
Department of Pediatric Surgery, All India Institute of Medical Sciences, New Delhi, India
| Date of Web Publication | 4-Jan-2014 |
Correspondence Address:
Minu Bajpai
Department of Pediatric Surgery, All India Institute of Medical Sciences, New Delhi - 110 029
India
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.124374

 Abstract Abstract | | |
Objective: The Objective of this study was to identify the association of mutation of fibroblast growth factor receptor 1 (FGFR1), FGFR2 genes with syndromic as well as non-syndromic craniosynostosis in Indian population.
Materials and Methods: Retrospective analysis of our records from January 2008 to December 2012 was done. A total of 41 cases satisfying the inclusion criteria and 51 controls were taken for the study. A total volume of 3 ml blood from the patient as well as parents was taken. Deoxyribonucleic acid extracted using phenol chloroform extraction method followed by polymerase chain reaction-restriction fragment length polymorphism method.
Results: There were 33 (80.4%) non-syndromic cases of craniosynostosis while 8 (19.5%) were syndromic. Out of these 8 syndromic cases, 4 were Apert syndrome, 3 were Crouzon syndrome and 1 Pfeiffer syndrome. Phenotypically the most common non-syndromic craniosynostosis was scaphocephaly (19, 57.7%) followed by plagiocephaly in (14, 42.3%). FGFR1 mutation (Pro252Arg) was seen in 1 (2.4%) case of non-syndromic craniosynostosis while no association was noted either with FGFR1 or with FGFR2 mutation in syndromic cases. None of the control group showed any mutation.
Conclusion: Our study proposed that FGFR1, FGFR2 mutation, which confers predisposition to craniosynostosis does not exist in Indian population when compared to the western world.
Keywords: Craniosynostosis, fibroblast growth factor receptor, non-syndromic, suture, syndromic
How to cite this article:
Pandey RK, Bajpai M, Ali A, Gayan S, Singh A. Mutational identification of fibroblast growth factor receptor 1 and fibroblast growth factor receptor 2 genes in craniosynostosis in Indian population. Indian J Hum Genet 2013;19:449-53 |
How to cite this URL:
Pandey RK, Bajpai M, Ali A, Gayan S, Singh A. Mutational identification of fibroblast growth factor receptor 1 and fibroblast growth factor receptor 2 genes in craniosynostosis in Indian population. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:449-53. Available from: http://www.ijhg.com/text.asp?2013/19/4/449/124374 |
| Introduction | |  |
The majority of known genetic causes of craniosynostosis are mutations in the genes encoding fibroblast growth factor receptor types 1-3 (FGFR 1, 2 and 3); [1] other significant genes are TWIST1 and EFNB1. A major breakthrough in understanding the genetic background of craniosynostosis has been the identification of genetic defects in several syndromes, including the most common Crouzon syndrome and Apert syndrome. In general, more than 40 genes and several variants were reported in the literature as genetic risk factors for craniosynostosis and only 60% heritability was explained. The presence of mutations in the group of genes coding for the FGFR in patients with Apert and Crouzon syndromes is now clearly established. [2],[3],[4] These genes code for receptors on the cell surface, which mediate the effects of fibroblast growth factors (FGF). The effects of FGFs are not fully understood, but they are already clearly implicated in important cellular processes such as cell growth, differentiation, migration and survival. Although 4 different genes are located in different chromosomes, the receptor proteins they encode for being very similar structurally. A number of craniosynostotic disorders have recently been ascribed to mutations in genes coding for FGFR 1, 2 and 3. [5] The common features of these FGFR associated conditions are the unilateral or bilateral premature ossification of the coronal suture. The present finding points out the importance, from both diagnostic and prognostic points of view, of early FGFR mutational screening in craniosynostotic conditions, even in forms that apparently do not involve closure of the coronal suture at birth.
Aim and objective
The aim is to identify association of mutation of FGFR1, FGFR2 genes with syndromic as well as non-syndromic craniosynostosis in Indian population.
| Materials and Methods | | |
Retrospective analysis of records of patients registered in craniosynostosis clinic from January 2008 to December 2012 was done. Ethical clearance from the institute's ethical committee was taken. Diagnosed cases of syndromic and non-syndromic craniosynostosis patients between 6 months and 12 years of age either pre-operative or postoperative were included in the study. Patients with primary microcephaly (secondary craniosynostosis), postural plagiocephaly, incomplete data and lost to follow-up were excluded from the study.
Diagnostic investigations include clinical examination and plain X-ray skull (anteroposterior, lateral and Towne's view) and non-contrast computed tomography with 3D reconstruction if required. Out of 63 registered cases, 41 satisfying the inclusion were taken for the study. Blood sample (3 ml) was taken from both the parents along with the child in ethylene-diamine-tetra-acetic acid vial. For control 50 healthy children of comparable age group, belonging to the same geographical region were included in this study.
Genomic deoxyribonucleic acid (DNA) was extracted from peripheral blood lymphocytes by phenol chloroform extraction method. [6],[7] Primers to diagnose common FGFR1 and FGFR2 mutations in this study are listed in [Table 1]. Custom-synthesized primers for FGFR1 and FGFR2 gene were designed (Sigma Aldrich Chemicals Pvt. Ltd., Bangalore, India). polymerase chain reaction (PCR) for each sample was performed in 0.2 ml, thin-walled tubes using 20 ng of DNA, 2-5 pmol of each primer, 200 mm dinucleotide triphosphates, 10 × PCR buffer, 1.5 mm MgCl 2 and 0.5 units of DyNAzyme II DNA Polymerase (Thermo Scientific). The PCR reaction was carried out in a T-100 DNA Engine (Bio-Rad, Hercules, CA, USA) Thermal cyclers under the following conditions: 95°C for 3 min, 35 cycles at 95°C for 30 s, annealing temperature as in [Table 1] for 30 s and 72°C for 1 min/KB and a final extension at 72°C for 7 min. Amplicons size were verified by gel electrophoresis by running the PCR product on 2% agarose gel with the 100 bp maker (ladder). After successful amplification, PCR products were digested as per the manufacturer's instructions with the respective restriction endonucleases mentioned in [Table 2] and analyzed on an ethidium bromide-stained 3.0% agarose gel with 50 bp Ladder.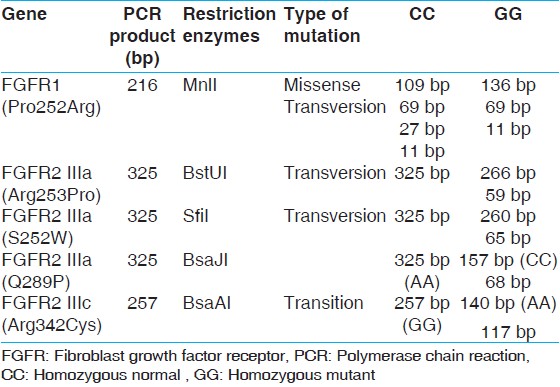 | Table 2: FGFR1 and FGFR2 amplicon size (bp) after PCR amplification and restriction digestion
Click here to view |
| Results | | |
There were 33 (80.4%) non-syndromic while 8 (19.5%) syndromic cases. Out of these 8 syndromic cases, 4 were Apert, 3 were Crouzon and 1 Pfeiffer. Phenotypically the most common non-syndromic craniosynostosis was scaphocephaly (19, 57.7%) followed by plagiocephaly in 14 (42.3%). FGFR1 mutation (Pro252Arg) was seen in 1 (2.4%) case of non-syndromic craniosynostosis while no association of mutation with either FGFR1 or FGFR2 mutation was noted in syndromic cases. The only mutation FGFR1 (Pro252Arg) seen, was in a boy with non-syndromic unicoronal craniosynostosis. The mutation was also present in her mother who had mild facial asymmetry, but do not have craniosynostosis. None of the control group showed any mutation. Our results suggest that the mutation is not associated with craniosynostosis with Indian children. [Figure 1] showing the PCR product amplification of FGFR1 on 2% Agarose gel, after successful amplification the product was digested. After digestion, all the fragments were resolved on 3% agarose gels. The homozygous normal allele (CC) produced 4 fragments of 109 bp, 69 bp, 27 bp, 11 bp size. The heterozygous (GC) produced 5 fragments of 109 bp, 136 bp, 69 bp, 27 bp, 11 bp size, whereas the homozygous mutant (GG) produced 3 fragments of 136 bp, 69 and 11 bp. However due to 69 bp, 27 bp, 11 bp are too small to be visualized so the normal, heterozygous and mutant allele were defined on the basis of bigger fragments, i.e. 109 and 136 bp. Lane 1 contains the PCR product of size 216 bp, Lane 2 contains the heterozygous condition (GG) size 109 and 136 bp for for craniosynostosis children and Lane 4, 5 shows digested PCR product of size 109 bp for non-craniosynostosis children [Figure 2].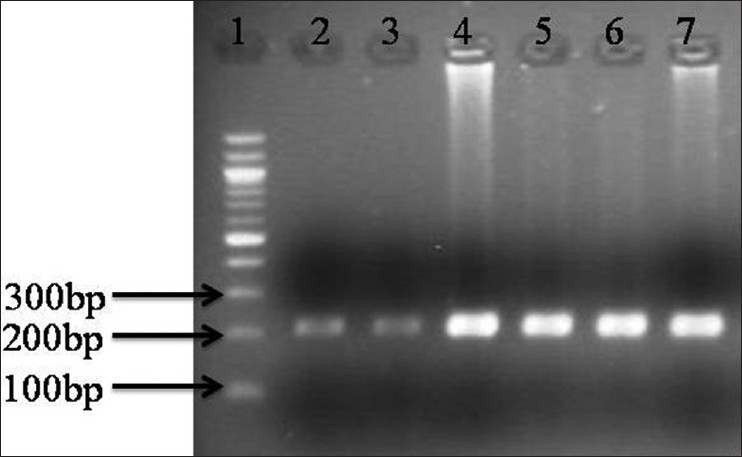 | Figure 1: 2% Agarose gel showing polymerase chain reaction (PCR) product of fibroblast growth factor receptor 1 amplification (216 bp) (1) Molecular maker (100 bp); (2-7) PCR prdocut of craniosynostosis and and non-craniosynostosis children
Click here to view |
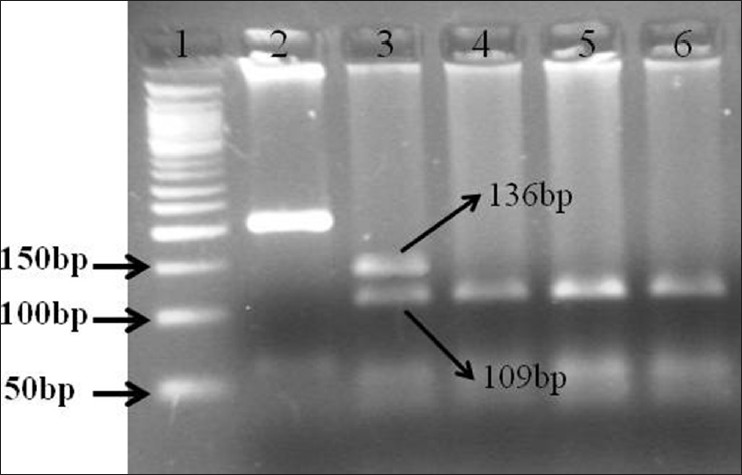 | Figure 2: Restriction fragment length polymorphism of polymerase chain reaction (PCR) product of fibroblast growth factor receptor 1 on 3% Agarose gel from craniosynostosis and non-craniosynostosis children's which were analysis for MnlI digestion. (1) Molecular marker (50 bp); (2) PCR product for craniosynostosis children's; (3) Completely digested PCR product for craniosynostosis children's (GC) (136 bp, 109 bp); (4 and 5) Completely digested PCR product for non-craniosynostosis children's (CC) (109 bp)
Click here to view |
| Discussion | | |
Children suffering from genetic disorders are not only social liability, but also an extra burden on the economy of developing countries such as India, Pakistan and Sri Lanka. Craniosynostosis is one of the major genetic disorders affecting the central nervous system in children with reported incidence of 1/2,200 live births. [8] First description of craniosynostosis was given by Otto in 1830. [9] Since then multiple theories have been proposed to explain the pathogenesis, with recent studies focusing on genetic regulation. [10] Still the etiology of the disease is largely unknown however the condition is related to abnormalities in the base of the skull and is frequently seen in association with osseous abnormalities of the face. Universally accepted hypothesis is an abnormal premature fusion of the cranial suture mainly because of an imbalance between proliferation and differentiation of cell. Craniosynostosis can either be non-syndromic or syndromic with the former more common than the latter. Various authors of the western world have reported the role of FGFR1 and 2 in craniosynostosis. [11],[12],[13],[14],[15] Our study is truly based on Indian population suffering from craniosynostosis. In English literature, multiple reports are available regarding the role of FGFR1 and FGFR2 genes in craniosynostosis, but similar data from the Indian subcontinent is still lacking. To the best of our knowledge, this is the pilot study from Asian subcontinent addressing the association of the FGFR1 and FGFR2 mutation with craniosynostosis. In the present study, we have screened the most common associated mutation of FGFR1 Pro252Arg [16],[17] and S252W in FGFR2, [18] Pro253Arg in FGFR2, [1],[2],[19],[20],[21],[22] Q289P, [23],[24] Arg342Cys [25],[26],[27] in Indian population. Pedigrees of 41 children suffering from both syndromic as well as non-syndromic craniosynostosis (Apert Syndrome, Crouzon and Pfeiffer syndromes) were analyzed. We did not find any association with FGFR2 mutation in families of children suffering from craniosynostosis. Only mutation (FGFR1 [Pro252Arg]) seen, was in a boy with non-syndromic unicoronal craniosynostosis. The mutation was also present in his mother who had mild facial asymmetry but do not have craniosynostosis. Age of the child was 32 months and on examination there was flattening of the frontal bone on the right side, with a prominent frontal eminence on the left side resulting in anterior plagiocephaly. There was no exorbitism, midface hypoplasia, oral palatal defect, or ear abnormality either in mother or son. There was no family history of craniosynostosis. The child doesn't show any peculiar features to differentiate him from others except the manifestation of underlying primary pathology. Most patients with craniosynostosis do not, however, have obvious syndromic features, making an accurate diagnosis and genetic counseling more difficult, particularly because non-syndromic craniosynostosis is likely to be etiologically heterogeneous. Up to 30% of such patients with coronal craniosynostosis have a specific mutation in FGFR3 (Pro250Arg) that is more reliably identified by genetic testing than by clinical features. Mutations of FGFR2 are much rarer in non-syndromic patients, but a small number of FGFR2 mutations have been identified in individuals with mild, atypical or more variable phenotypes. Our study provides evidence that the mutation is not associated with craniosynostosis in Indian children, although it has been shown to be associated with children in Australia and other western countries. Because India is known for its diversity and complexity of genome therefore, it is important to perform replicate studies of patients from diverse ethnic origins (different states of India) before either designating or excluding this mutation as a risk for craniosynostosis. This study will help in better understanding of the existing genetic mutations in Indian children suffering from this grave disease thereby providing an opportunity to the treating pediatric surgeons to reduce the agony and suffering of the children. The present study tries to establish a novel bimolecular marker for consideration and determination of craniosynostosis patients in India. Our study has its own limitation such as small sample size for arriving at any definitive conclusion. In a recent study, Wilkie et al. has shown the prevalence and complications of a single gene and chromosomal disorders in craniosynostosis. [28] They concluded that cytogenetic and molecular genetic testing, as a minimum for mutations in FGFR3 (P250R) and FGFR2 exons IIIa and IIIc, should be an integral part of management in children with bicoronal, unicoronal or multisuture synostosis. Research aimed at identifying new genetic mutation in craniosynostosis requires careful choice of the patients, as many with chromosomal abnormalities or other syndromes may have secondary causes. Other publications on craniosynostosis involving European ancestry (NHW) populations were important from the perspective of population specific understanding of the genetic causes. Justice et al. in their study of 130 non-syndromic cases showed the susceptibility loci for non-syndromic sagittal craniosynostosis near BMP2 and within BBS9 and was associated with familial (case-parent trios of European ancestry) craniosynostosis. It also represented the first major step toward deciphering the genetic etiology of non-syndromic sagittal craniosynostosis (sins). [29] Yagnik et al. reported that ALX4 variants may have an impact on the genetic etiology of non-syndromic craniosynostosis. [30] Seto et al., in a series of 164 cases have shown that genetic testing of patients with isolated sagittal or coronal synostosis should include TWIST1 mutational analysis. [31] Study of Asian continent through Korea by Yu et al. showed the genotypic and phenotypic analyses of Korean patients with syndromic craniosynostosis. [32]
| Conclusion | | |
Our study provides the strongest evidence that association of mutation of FGFR1, FGFR2 with syndromic as well as non-syndromic craniosynostosis does not exist in Indian population as seen in western population. Our study will provide the necessary platform for further research to better understand the genomics of craniosynostosis in Indian population.
| References | | |
| 1. | Passos-Bueno MR, Sertié AL, Zatz M, Richieri-Costa A. Pfeiffer mutation in an Apert patient: How wide is the spectrum of variability due to mutations in the FGFR2 gene? Am J Med Genet 1997;71:243-5. 
|
| 2. | Hollway GE, Suthers GK, Haan EA, Thompson E, David DJ, Gecz J, et al. Mutation detection in FGFR2 craniosynostosiss yndromes. Hum Genet 1997;99:251-5.
|
| 3. | Von Gernet S, Golla A, Ehrenfels Y, Schuffenhauer S, Fairley JD. (2000) Genotype-phenotype analysis in Apert syndrome suggests opposite effects of the two recurrent mutations on syndactyly and outcome of craniofacial surgery. Clin Genet.57:137-9.
|
| 4. | Lajeunie E, Heuertz S, El Ghouzzi V, Martinovic J, Renier D, Le Merrer M, Bonaventure J. (2006) Mutation screening in patients with syndromic craniosynostoses indicates that a limited number of recurrent FGFR2 mutations accounts for severe forms of Pfeiffer syndrome. Eur J Hum Genet. 14:289-98.
|
| 5. | Tartaglia M, Bordoni V, Velardi F, Basile RT, Saulle E, Tenconi R, Di Rocco C, Battaglia PA. (1999) Fibroblast growth factor receptor mutational screening in newborns affected by metopic synostosis. Childs Nerv Syst.15:389-93.
|
| 6. | Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning a Laboratory Manual. 2 nd ed. New York: Cold Spring Harbor Laboratory; 1989.
|
| 7. | Thangaraj K, Joshi MB, Reddy AG, Gupta NJ, Chakravarty B, Singh L. CAG repeat expansion in the androgen receptor gene is not associated with male infertility in Indian populations. J Androl 2002;23:815-8.
|
| 8. | Reefhuis J, Honein MA, Shaw GM, Romitti PA. Fertility treatments and craniosynostosis: California, Georgia, and Iowa, 1993-1997. Pediatrics 2003;111:1163-6.
|
| 9. | Otto W. A Berlin: Ruecker, in 1830. Textbook of pathological anatomy and the beasts of the meuchen. Vol. 1. Berlin: Ruecker; 1830.
|
| 10. | Persing JA, Jane JA, Shaffrey M. Virchow and the pathogenesis of craniosynostosis: A translation of his original work. Plast Reconstr Surg 1989;83:738-42.
|
| 11. | Mulliken JB, Steinberger D, Kunze S, Muller U.(1999) Molecular diagnosis of bilateral coronal synostosis. Plast Reconstr Surg. 104:1603-15.
|
| 12. | Mulliken JB, Gripp KW, Stolle CA, Steinberger D, Muller U. (2004) Molecular analysis of patients with synostotic frontal plagiocephaly (unilateral coronal synostosis). Plast Reconstr Surg. 113:1899-909.
|
| 13. | White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, McKeown C, Fitzpatrick D, Yu K, Ornitz DM, Econs MJ. (2005) Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet.76:361-7.
|
| 14. | Hackett A, Rowe L. (2006) FGFR1 Pfeiffer syndrome without craniosynostosis: An additional case report. Clin Dysmorphol. 15:207-10.
|
| 15. | Farrow EG, Davis SI, Mooney SD, Beighton P, Mascarenhas L, Gutierrez YR, Pitukcheewanont P, White KE. (2006) Extended mutational analyses of FGFR1 in osteoglophonic dysplasia. Am J Med Genet A. 140:537-9.
|
| 16. | Muenke M, Schell U, Hehr A, Robin NH, Losken HW, Schinzel A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet 1994;8:269-74.
|
| 17. | Meyers GA, Day D, Goldberg R, Daentl DL, Przylepa KA, Abrams LJ, et al. FGFR2 exon IIIa and IIIc mutations in Crouzon, Jackson-Weiss, and Pfeiffer syndromes: Evidence for missense changes, insertions, and a deletion due to alternative RNA splicing. Am J Hum Genet 1996;58:491-8.
|
| 18. | 18 Moloney DM, Slaney SF, Oldridge M, Wall SA, Sahlin P, Stenman G, et al. Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet 1996;13:48-53.
|
| 19. | Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet 1995;9:165-72.
|
| 20. | Park WJ, Theda C, Maestri NE, Meyers GA, Fryburg JS, Dufresne C, et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet 1995a;57:321-8.
|
| 21. | Slaney SF, Oldridge M, Hurst JA, Moriss-Kay GM, Hall CM, Poole MD, et al. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet 1996;58:923-32.
|
| 22. | Filkins K, Russo JF, Boehmer S, Camous M, Przylepa KA, Jiang W, et al. Prenatal ultrasonographic and molecular diagnosis of Apert syndrome. Prenat Diagn 1997;17:1081-4.
|
| 23. | Freitas EC, Nascimento SR, de Mello MP, Gil-da-Silva-Lopes VL. Q289P mutation in FGFR2 gene causes Saethre-Chotzen syndrome: Some considerations about familial heterogeneity. Cleft Palate Craniofac J 2006;43:142-7.
|
| 24. | Piccione M, Antona V, Niceta M, Fabiano C, Martines M, Bianchi A, et al. Q289P mutation in the FGFR2 gene: First report in a patient with type 1 Pfeiffer syndrome. Eur J Pediatr 2009;168:1135-9.
|
| 25. | Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet 1994;8:98-103.
|
| 26. | Rutland P, Pulleyn LJ, Reardon W, Baraitser M, Hayward R, Jones B, et al. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nat Genet 1995;9:173-6.
|
| 27. | Park WJ, Meyers GA, Li X, Theda C, Day D, Orlow SJ, et al. Novel FGFR2 mutations in Crouzon and Jackson-Weiss syndromes show allelic heterogeneity and phenotypic variability. Hum Mol Genet 1995b;4:1229-33.
|
| 28. | Wilkie AO, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics 2010;126:e391-400.
|
| 29. | Justice CM, Yagnik G, Kim Y, Peter I, Jabs EW, Erazo M, et al. A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal Craniosynostosis near BMP2 and within BBS9. Nat Genet 2012;44:1360-4.
|
| 30. | Yagnik G, Ghuman A, Kim S, Stevens CG, Kimonis V, Stoler J, et al. ALX4 gain-of-function mutations in nonsyndromic craniosynostosis. Hum Mutat 2012;33:1626-9.
|
| 31. | Seto ML, Hing AV, Chang J, Hu M, Kapp-Simon KA,Patel PK, et al. Isolated sagittal and coronal Craniosynostosis associated with TWIST box mutations. Am J Med Genet A 2007;143:678-86.
|
| 32. | Yu JE, Jeong SY, Yang JA, Park MS, Kim HJ,Yoon SH. Genotypic and phenotypic analyses of Korean patients with syndromic craniosynostosis. Clin Genet 2009;76:287-91.
|
[Figure 1], [Figure 2]
[Table 1], [Table 2]
|