|
Show all abstracts Show selected abstracts Add to my list |
|
| EDITORIALS |
|
|
|
Neural tube defects: A need for population-based prevention program |
p. 145 |
Shubha Phadke, Meenal Agarwal
DOI:10.4103/0971-6866.100747 PMID:23162285 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Micro mapping the frequencies of beta thalassemia and sickle cell anemia in India: A way forward to plan control strategies |
p. 148 |
Reena Das
DOI:10.4103/0971-6866.100748 PMID:23162286 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| REVIEW ARTICLES |
 |
|
|
|
Stem cells: A potential regenerative future in dentistry |
p. 150 |
Sumit Narang, Nidhi Sehgal
DOI:10.4103/0971-6866.100749 PMID:23162287In recent years, the field of dentistry has embossed its presence by taking major leaps in research and further bringing it into practice. The most valuable ongoing research in regenerative dentistry is the study on stem cells. It was instituted that stem cells grow rapidly and have the potential to form specialized dentin, bone, and neuronal cells. These neuronal cells can be used for dental therapies and can provide better treatment options for patients. The stem cells based therapies could help in new advances in treating damaged teeth, inducing bone regeneration and treating neural injury as well. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Toll-like receptors in autoimmunity with special reference to systemic lupus erythematosus |
p. 155 |
Vandana D Pradhan, Swaptagni Das, Prathamesh Surve, Kanjaksha Ghosh
DOI:10.4103/0971-6866.100750 PMID:23162288The Toll-like receptor (TLR) family plays a fundamental role in host innate immunity by mounting a rapid and potent inflammatory response to pathogen infection. TLRs recognize distinct microbial components and activate intracellular signaling pathways that induce expression of host inflammatory genes. Several studies have indicated that TLRs are implicated in many inflammatory and immune disorders. Extensive research in the past decade to understand TLR-mediated mechanisms of innate immunity has enabled pharmaceutical companies to begin to develop novel therapeutics for the purpose of controlling an inflammatory disease. The roles of TLRs in the development of autoimmune diseases have been studied. TLR7 and TLR9 have key roles in production of autoantibodies and/or in development of systemic autoimmune disease. It remains to be determined their role in apoptosis, in the pathogenesis of RNA containing immune complexes, differential expression of TLRs by T regulatory cells. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| ORIGINAL ARTICLES |
|
|
|
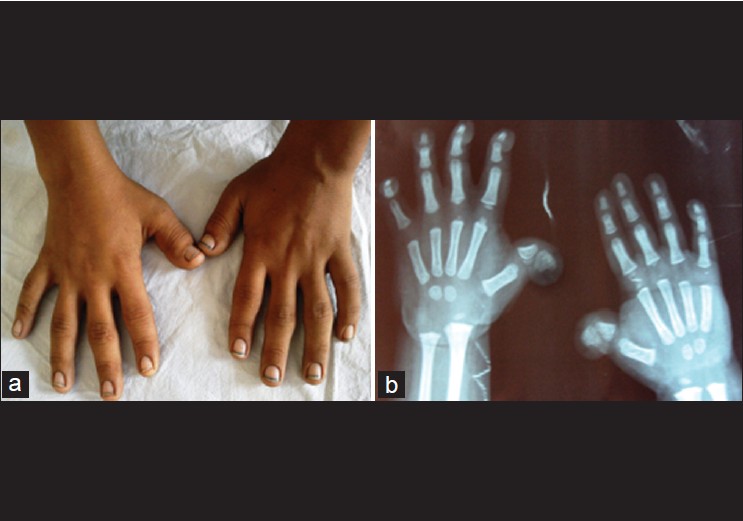  |
Rubinstein-taybi syndrome: Clinical profile of 11 patients and review of literature |
p. 161 |
Suresh Kumar, Renu Suthar, Inusha Panigrahi, Ram K Marwaha
DOI:10.4103/0971-6866.100751 PMID:23162289Background: Rubinstein-Taybi syndrome (RSTS) is a rare congenital neurodevelopmental disorder, characterized by postnatal growth deficiency, typical dysmorphic features, broad thumbs and toes, and mental retardation. Very few cases are reported in literature from developing countries. Diagnosis is often delayed due to non-familiarity with the characteristic features of this syndrome.
Aims: To report 11 cases of RSTS and to review the current literature.
Settings And Design: Retrospective study conducted in genetic and metabolic unit of a tertiary care teaching hospital in north India over a period of 3½ years. Materials And Methods: 11 patients with diagnosis of RSTS were identified, and their case sheets were reviewed.
Results: Developmental delay was presenting complaint in 10 patients, and seizure in 1 case. 7 patients had microcephaly (head circumference below −3 SD), and a prominent beaked nose was seen in 9 patients. The intelligence quotient (IQ) varied from 22-62 in 7 patients who had mental retardation. The most notable features in hands were broadness, shortening, and flattening of the distal phalanx of thumbs or great toes. Additionally, we also noted webbing of neck, microphthalmia, and pachygyria (on MRI brain) in 1 patient each.
Conclusions: The diagnosis of RSTS is primarily clinical and based on characteristic phenotype that is often combined with a variety of somatic anomalies. An early diagnosis facilitates appropriate genetic counseling and in planning the management. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (4) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Prevalence and hematological profile of β-thalassemia and sickle cell anemia in four communities of Surat city |
p. 167 |
Dipal S Bhukhanvala, Smita M Sorathiya, Avani P Shah, Ankur G Patel, Snehalata C Gupte
DOI:10.4103/0971-6866.100752 PMID:23162290Background: From the data of transfusion-dependent thalassemia major cases, the 4 communities (Muslim, Dhodia Patel, Kachhiya Patel, and Modh Bania) with high prevalence but not studied methodically were selected.
Aim: The aim of this study is to find prevalence of β-thalassemia and sickle cell anemia in 4 selected communities and also to evaluate hematological profile in them.
Materials and Methods: For screening of β-thalassemia trait (BTT) and sickle cell trait (SCT), all samples were tested for red cell indices, solubility, HbA 2 level and doubtful cases confirmed on HPLC.
Statistical Analysis: Mean ± SD, χ2 and 't' tests were used to evaluate the significance.
Results and Conclusion: Among 4 selected communities, the highest prevalence of BTT was observed in Modh Bania (6.2%) and Kachhiya Patel (6.05%) and that of SCT in Dhodia Patel (14.0%). Significantly higher prevalence of BTT was observed in Memon ( P < 0.0001) and of SCT in Khalifa 6.6% ( P < 0.0001) compared to other Muslim sub castes. Anemia was more prevalent in BTT compared to non-BTT and non-SCT subjects. 80% of Dhodia Patel non-BTT and non-SCT subjects showed microcytic red cell morphology. Their Mean ± SD Hb concentration was 12.1 ± 1.73, hence iron deficiency cannot be a sole reason. This community needs α-thalassemia and iron studies. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (4) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
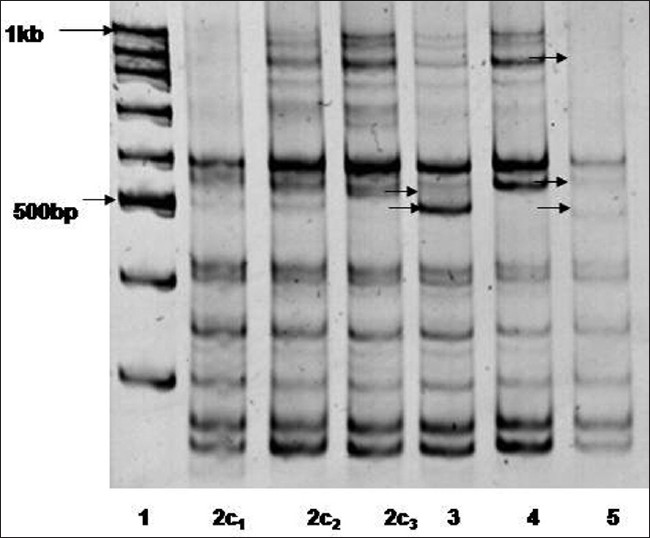 |
Association of microsatellite instability and chronic obstructive pulmonary disorder in isocyanate-exposed population of Bhopal |
p. 172 |
Protiti Bose, Rashmi Bathri
DOI:10.4103/0971-6866.100754 PMID:23162291Context: Survivors of the Bhopal gas disaster still suffer from various respiratory ailments. We examined the effects of exposures among a cross-section of current residents suffering from COPD by ISSR-PCR.
Aims: Molecular screening of the gas-affected population of Bhopal with COPD for microsatellite instability due to exposure of MIC.
Settings and Design: The isocyanate-exposed population of Bhopal city suffering from chronic obstructive pulmonary disorder.
Materials and Methods: Inter-(SSR) analysis was used to characterize microsatellite instability in 52 MIC victims of Bhopal, suffering from COPD using (CA) 8 RG and (CA) 8 R[Y-Q] primer.
Statistical Analysis Used: Association analyses were performed using regression analysis.
Results: The study on the MIC-affected population in Bhopal showed weak association between microsatellite instability and age (r = + 0.37); exposure distance from site (r = −0.44); and smoking status(r = + 0.12); while regression analysis of the above parameters displayed supporting evidence.
Conclusions: The high prevalence of smoking coupled with aging and poor living habits threatens, to further increase COPD incidences among this population, highlighting the need for enhanced screening efforts. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
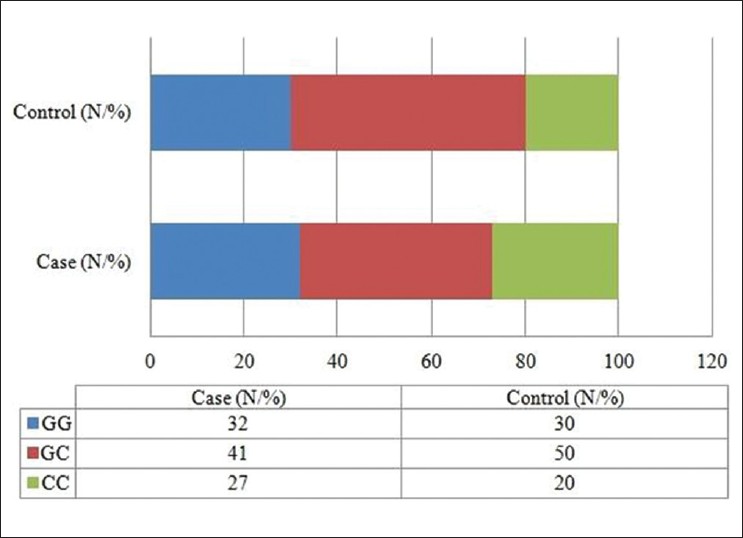 |
Effect of maternal Tp53 gene G412C polymorphism on neural tube defects: A study from North India |
p. 177 |
Jyoti Arora, Kallur N Saraswathy, Roumi Deb
DOI:10.4103/0971-6866.100757 PMID:23162292Context: Tumor protein 53 (tp53) is one of the candidate gene proposed for neural tube defects, which affects central nervous system during early embryonic development, on the basis of mouse models.
Aims: The present study is an attempt to unfold the possible role of tp53 G412C polymorphism in the incidence of neural tube defect (NTDs) in humans.
Settings and Design: Case-control study was carried out in government hospitals of Delhi, India.
Materials and Methods: Subjects comprised of 100 mothers of NTD children and 100 matched control mothers. Information on some environmental exposures was collected along with blood samples. After DNA extraction, the genotyping of tp53 G412C polymorphism was carried out by PCR-RFLP method.
Statistical Analysys: Fisher Exact or Chi square test, binary logistic model, and odds ratio (95% confidence interval) calculations were used to evaluate effect of risk factors on NTDs using SPSS v17.0.
Results: The 'CC' genotype of tp53 G412C showed protective effect towards the development of anencephaly and/or encephalocele (OR: 0.44; 95% CI: 0.19-1.00); however, no significant difference among overall NTD cases and controls was observed (P>0.05). Further segregation of all subjects based on 2 different communities, Hindus and Muslims, the association of 'CC' genotype of the polymorphism with reduced NTD risk was observed among Hindu community (OR: 0.33; 95% CI: 0.13-0.79).
Conclusion: The study highlights the selective advantage provided by maternal 'CC' genotype, thereby reducing risk of cephalic NTDs, probably due to the lower apoptotic activity of the protein, however, more specifically in the presence of community-specific microenvironment. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
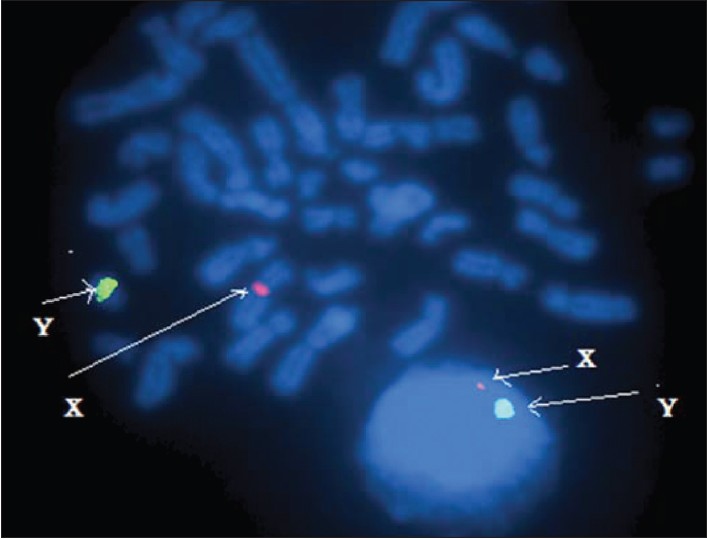 |
Chromosomal abnormalities and hormonal disorders of primary amenorrhea patients in Egypt |
p. 183 |
Faeza El-Dahtory
DOI:10.4103/0971-6866.100758 PMID:23162293Background: Primary amenorrhea is defined as the absence of menstruation and secondary sexual characteristics in phenotypic women aged 14 years or older. Hormonal disorders are main causes of primary amenorrhea. Common hormonal cause of primary amenorrhea includes pituitary dysfunction and absent ovarian function. The aim of this study was to estimate the incidence and types of chromosomal abnormalities in patients with primary amenorrhea in Egypt.
Materials and Methods: Chromosomal analysis and hormonal assay were carried out on 223 patients with primary amenorrhea that were referred from different parts of Egypt to Cytogenetic laboratory of Genetic Unit, Children Hospital Mansoura University, from July 2008 to December 2010. FISH technique was carried out in some of cases to more evaluation.
Results: The frequency of chromosomal abnormalities was 46 (20.63%) in primary amenorrhea patients. The chromosomal abnormalities can be classified into four main types. (1) The numerical abnormalities of the X chromosome were detected in 23 (50 %). (2) Structural abnormalities of the X chromosome were detected in 11 (23.91%). (3) Mosaicism of X chromosome was found in 10 (21.74%). (4) Male karyotype 46, XY was presented in 2 (4.35%).
Conclusion: The present study showed that karyotype and FISH are necessary to detect the causes of primary amenorrhea. This study also revealed the incidence of chromosomal abnormalities in women with primary amenorrhea in Egypt is similar to that reported in previous literatures. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
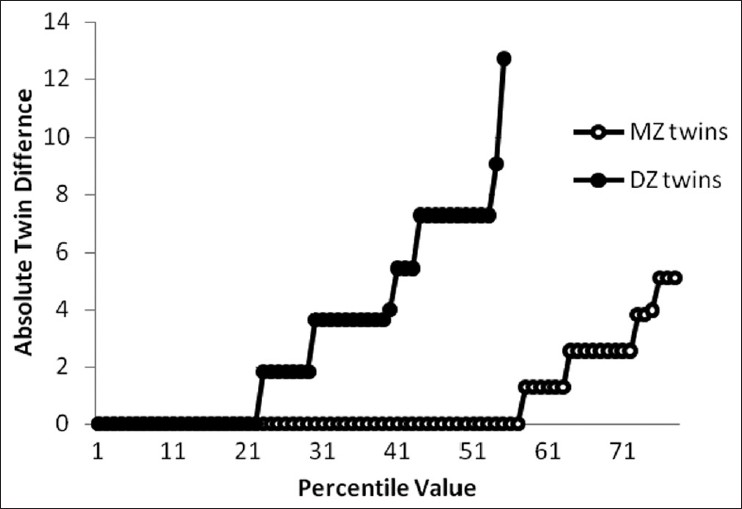 |
Genetic and environmental determinants of menstrual characteristics |
p. 187 |
Shayesteh Jahanfar
DOI:10.4103/0971-6866.100759 PMID:23162294Background: The impact of women's menstrual cycle on her quality of life, health, work, and community is substantial. Menstrual disturbance is linked with general ill conditions such as migraine, asthma, and endocrinopathies. The clinical significance of medical interventions to prevent these conditions becomes clear if the role of genetic or environment is clarified.
Aims: To identify the genetic and environmental contribution on menstrual characteristics.
Setting and Design: This was a cross-sectional study in 2 Asian countries.
Materials and Methods: 2 cohorts of monozygotic and dizygotic twins born between (1945-1988, n = 122) and (1951-1993, n = 71) were taken. A standard questionnaire was designed inclusive of socio- demographic characteristics of subjects as well as menstrual history (duration, interval, amount, irregularity). Subjects were interviewed by phone.
Statistical Analysis: Quantitative variables were analyzed using Falconars' formula as well as maximum likelihood analysis. Structural modeling was then applied to twin correlations to provide estimates of the relative genetic and/or environmental factors contribution in determining the measured trait.
Results: Menstrual characteristics were found to be under environmental influence where the best fitting model for menstrual interval and duration was common environment. CDF plotting confirmed the results for both variables. Proband-wise concordance analysis for amount of menstruation, amenorrhea, and irregular menstruation revealed no genetic influence. The best fitting model for menstrual irregularity was CE (C73%, E27%). The same model was defined for amenorrhea (C48%, E52%).
Conclusions: Environmental factors are most likely responsible to determine the menstrual flow, its integrity, and regularity. These factors need to be studied further. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (3) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
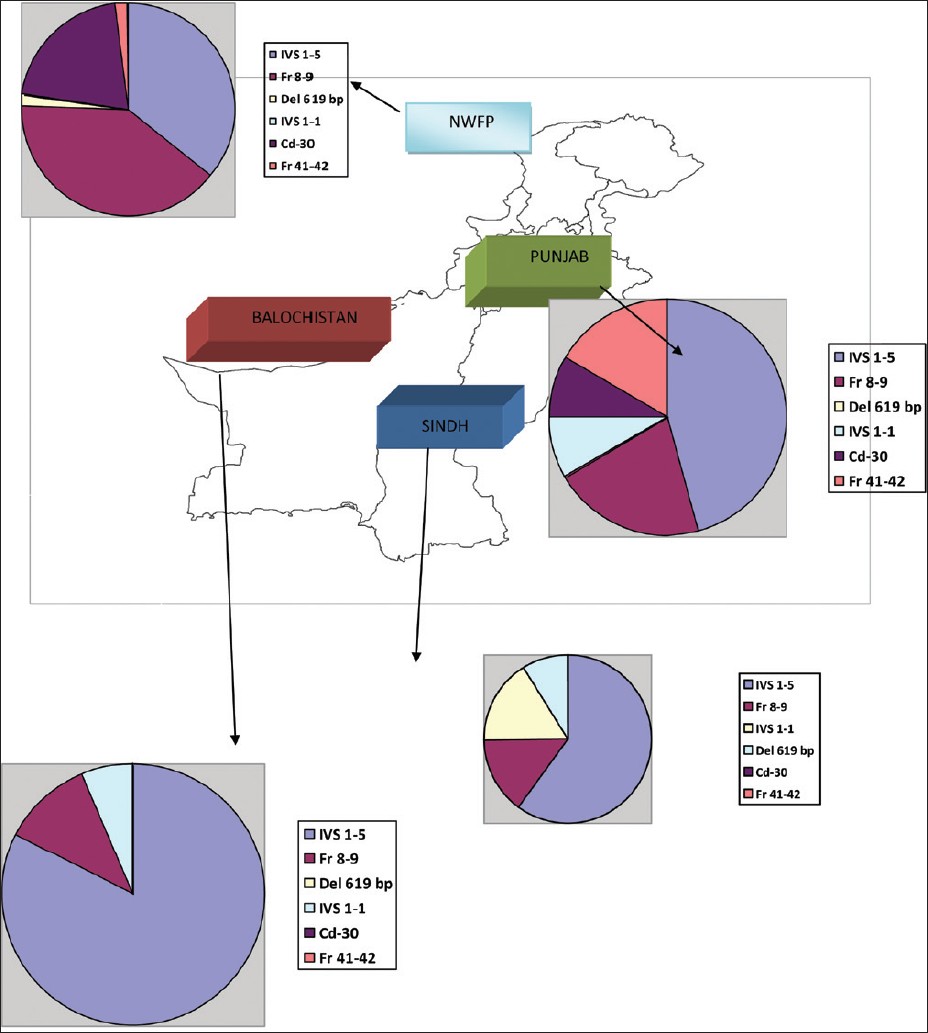 |
Molecular epidemiology of β-thalassemia in Pakistan: Far reaching implications |
p. 193 |
Saqib H Ansari, Tahir S Shamsi, Mushtaq Ashraf, Tasneem Farzana, Muneera Bohray, Kousar Perveen, Sajida Erum, Iqra Ansari, Muhammad Nadeem Ahmed, Masood Ahmed, Faizan Raza
DOI:10.4103/0971-6866.100762 PMID:23162295Background: β -Thalassaemia, an autosomal recessive hemoglobinopathy, is one of the commonest genetically transmitted disorders throughout the world. Collective measures including carrier identification, genetic counseling and prenatal diagnosis are required for preventing β-thalassemia.
Aim: To achieve this objective, Identification of the spectrum of genetic mutations, especially for various ethnic backgrounds in Pakistan. Therefore, we designed a cross sectional prospective study to identify the frequency of various gene mutations in different ethnic groups of Pakistan.
Materials and Methods: Over a 5-year period, DNA from 648 blood samples {including specimens of chorionic villus sampling (CVS)} were analyzed for the twelve most common β-thalassemia mutations found in the Pakistani population by a Multiplex amplification refractory mutation system (ARMS). Each sample was analyzed for the mutation as well as the normal gene, appropriate with negative and positive controls, and reagent blanks.
Results: Out of 648 samples mutations were identified in 640 (98.75%) samples by multiplex ARMS. 8 common β-thalassemia mutations were identified in 8 different ethnic groups accounting for 93.9% of the β-thalasemia alleles.
Conclusions: Based on the outcome of this study a cost effective proposal is formulated for detection of β-thalassemia mutations. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Cytogenetic abnormalities in 222 infertile men with azoospermia and oligospermia in Iran: Report and review |
p. 198 |
Mohammad T Akbari, F Behjati, GR Pourmand, F Akbari Asbagh, M Ataei Kachoui
DOI:10.4103/0971-6866.100764 PMID:23162296Background: Infertility affects approximately 10%-15% of couples in reproductive age. In half of the couples, causes are male-related, associated with impaired spermatogenesis. There is a complex correlation between genetics and infertility. Several factors affect on gametogenesis, from which factors that lead to chromosomal abnormalities are one of the best known. The aim of this study was to determine type and rate of chromosomal abnormalities in infertile azoospermic and oligospermic males in Iranian population.
Materials and Methods: The records of a total of 222 participants were evaluated retrospectively.
Results: As a whole we observed 13.96% chromosomal abnormality, from which 12.15% showed numerical and 1.8% showed structural abnormalities.
Conclusion: Comparison of our results with the review of the literature shows a higher incidence (4- fold) of gonosomal, in particular, numerical gonosomal, chromosomal anomalies. Cytogenetic analysis is strongly suggested for infertile men, particularly in those who suffer from azoospermia. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
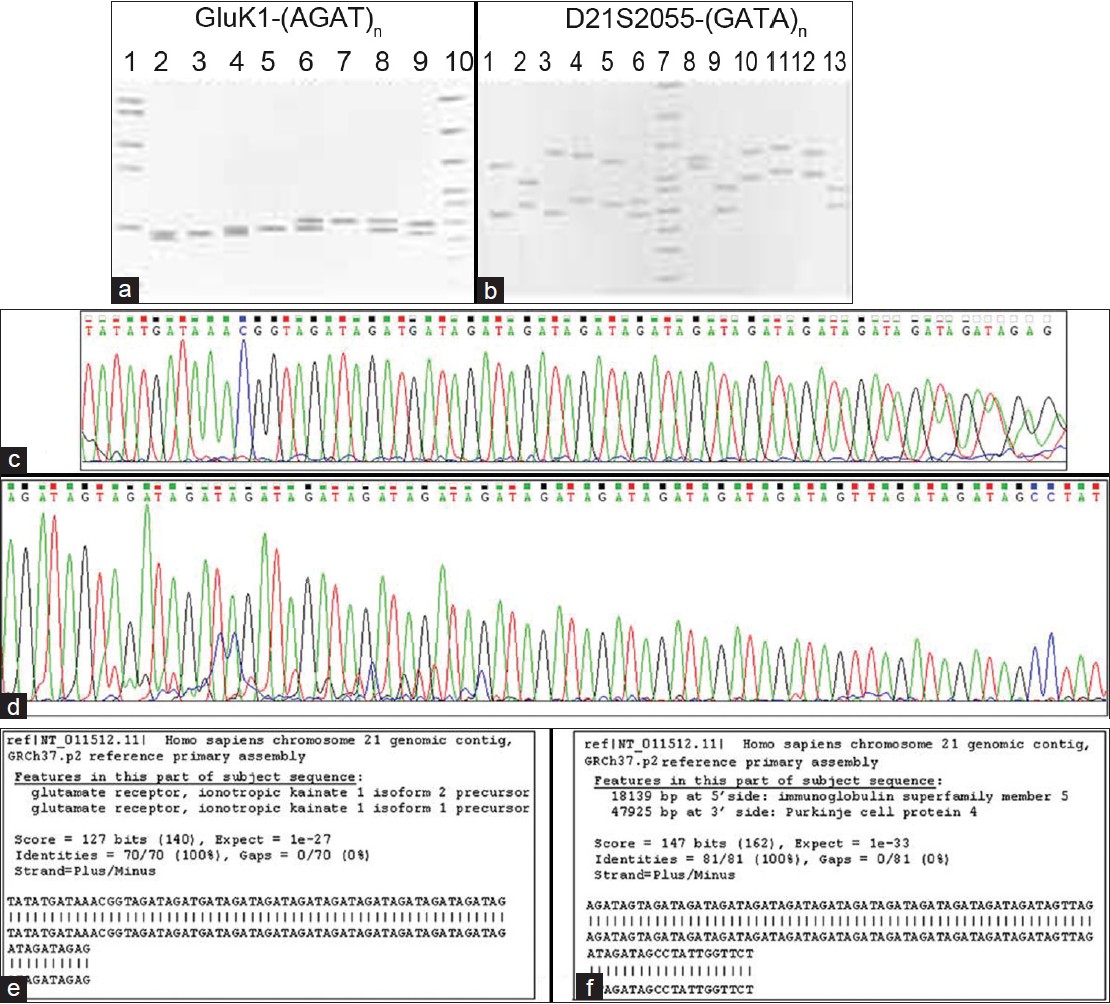 |
Discerning non-disjunction in down syndrome patients by means of GluK1-(AGAT) n and D21S2055-(GATA) n microsatellites on chromosome 21 |
p. 204 |
Ghosh Debarati, Sinha Swagata, Chatterjee Anindita, Nandagopal Krishnadas
DOI:10.4103/0971-6866.100769 PMID:23162297Introduction: Down syndrome (DS), the leading genetic cause of mental retardation, stems from non-disjunction of chromosome 21.
Aim: Our aim was to discern non-disjunction in DS patients by genotyping GluK1-(AGAT) n and D21S2055-(GATA) n microsatellites on chromosome 21 using a family-based study design.
Materials and Methods: We have used a PCR and automated DNA sequencing followed by appropriate statistical analysis of genotype data for the present study
Results and Discussion: We show that a high power of discrimination and a low probability of matching indicate that both markers may be used to distinguish between two unrelated individuals. That the D21S2055-(GATA) n allele distribution is evenly balanced, is indicated by a high power of exclusion [PE=0.280]. The estimated values of observed heterozygosity and polymorphism information content reveal that relative to GluK1-(AGAT) n [H obs =0.286], the D21S2055- (GATA) n [H obs =0.791] marker, is more informative. Though allele frequencies for both polymorphisms do not conform to Hardy-Weinberg equilibrium proportions, we were able to discern the parental origin of non-disjunction and also garnered evidence for triallelic (1:1:1) inheritance. The estimated proportion of meiosis-I to meiosis-II errors is 2:1 in maternal and 4:1 in paternal cases for GluK1-(AGAT) n , whereas for D21S2055-(GATA) n , the ratio is 2:1 in both maternal and paternal cases. Results underscore a need to systematically evaluate additional chromosome 21-specific markers in the context of non-disjunction DS. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
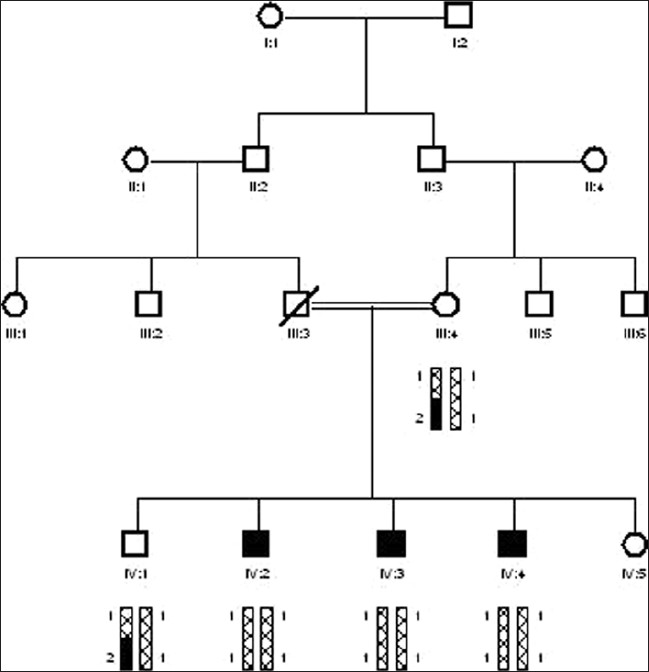 |
The study of gene GJB2/DFNB1 causing deafness in humans by linkage analysis from district Peshawar |
p. 217 |
Zafar Ali, Masroor Ellahi Babar, Jamil Ahmad, Sajjad Ali Shah
DOI:10.4103/0971-6866.100771 PMID:23162298Families with at least 2 or more individuals having hereditary hearing loss were enrolled from different areas of Khyber Pakhtoonkhwa, mainly from district Peshawar. Detailed history was taken from each family to minimize the presence of other abnormalities and environmental causes for deafness. Families were questioned about skin pigmentation, hair pigmentation, and problems relating to balance, vision, night blindness, thyroid, kidneys, heart, and infectious diseases like meningitis, antibiotic usage, injury, and typhoid. The pedigree structures were based upon interviews with multiple family members, and pedigrees of the enrolled families were drawn using Cyrillic program (version 2.1). All families showed recessive mode of inheritance. I studied 8 families of these 10. For linkage analyses, studies for DFNB1 locus, 3 STR markers (D13S175, D13S292, and D13S787) were genotyped using polyacrylamide gel electrophoresis (PAGE) and haplotypes were constructed to determined, linkage with DFNB1 locus. From a total of 8 families, a single family-10 showed linkage to DFNB1 locus. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Lack of association between the G-660C polymorphism in the dopamine transporter gene (SLC6A3) and schizophrenia in the Iranian population |
p. 222 |
Ali M Foroughmand, Hamid Galehdari, Bentalhoda Tirband Dastgerdi, Saeed Reza Khatami, Maryam Haidari
DOI:10.4103/0971-6866.100773 PMID:23162299Background: Dopaminergenic system plays an essential role in the plasticity of the human brain. The dopamine transporter gene (SLC6A3) mediates active reuptake of dopamine from synapsis, terminates dopamine signals, and therefore, is implicated in a number of dopamine-related disorders like psychosis. Variations in the form of single nucleotide polymorphisms in the core promoter of the SLC6A3 gene are reported to be involved in the pathogenesis of schizophrenia. In this study, we also attempted to establish the possible role of the polymorphism G-660C in the SLC6A3 gene promoter in schizophrenia in a case-control study.
Materials and Methods: The allele and genotype frequency were analyzed in an Iranian cohort of 200 unrelated patients and 200 controls using polymerase chain reaction and restriction fragment length polymorphism.
Results: The genotype frequency for case and control groups was GG 100%, GC 0%, CC 0%, and GG 100%, GC 0%, CC 0%, respectively. The C allele was failed in both groups.
Conclusion: Our data suggest clearly that there is no association between the -660G/C polymorphism and outcome of schizophrenia in the Iranian population. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Adjusted classification for ultrasound scoring index for antenatal detection of fetal trisomy |
p. 226 |
Viroj Wiwanitkit
DOI:10.4103/0971-6866.100775 PMID:23162300Ultrasound (USG) is a useful investigation in obstetrics. Its mean indications include screening for fetal anomaly, especially for Down's syndrome and other genetic trisomy. Here, the author tries to access the compatibility between classical USG scoring index and the new likelihood ratio-based system. New recommendation on severity of studied markers is given. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| BRIEF REPORT |
|
|
|
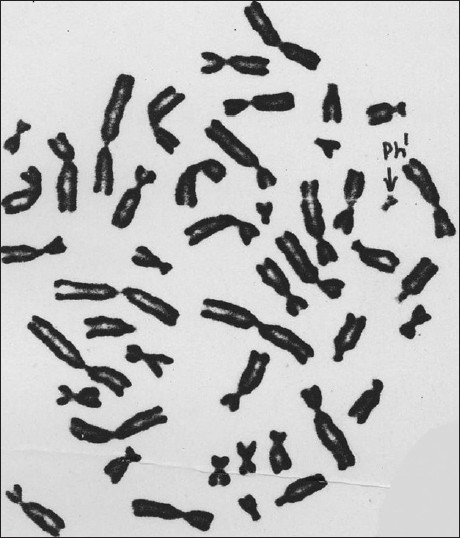 |
An investigation of Ph 1 chromosome in chronic myeloid leukemia patients with different treatment modalities and hematological features |
p. 229 |
Koushik Chattopadhyay, Bibhas Kar
DOI:10.4103/0971-6866.100778 PMID:23162301Chronic myeloid leukemia (CML) is characterized by a Ph 1 chromosome that derives through a translocation between chromosomes 9 and 22, i.e., t (9;22). Identifying the Ph 1 chromosome through cytogenetic analysis is an important aspect of CML diagnosis. The aim of this study was to determine the significance of cytogenetic analysis in the diagnosis of CML as well as to find out a relationship between chromosomal abnormalities and CML patients in different stages of treatment. Six CML patients were investigated for this study. The presence of Ph 1 chromosome was detected at different times of treatment using GTG banding on peripheral blood or bone marrow aspirations, and the results were analyzed using cytovision workstation. Hematological features were compared between newly diagnosed patients and patients under treatment. The Ph 1 chromosome was strongly associated with all cases of CML. The regression of Ph 1 chromosomes differed for each patient depending on the treatments and individual response to specific treatments. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| CASE REPORTS |
|
|
|
|
Crigler-Najjar syndrome type 2: Novel UGT1A1 mutation |
p. 233 |
Karippoth Mohandas Nair, Peter Lohse, Sheela Nampoothiri
DOI:10.4103/0971-6866.100776 PMID:23162302Crigler-Najjar syndrome type 2 is a rare cause for persistent unconjugated hyperbilirubinemia, inherited in an autosomal recessive manner. Even though it is compatible with normal life span, in the absence of prompt suspicion and intensive management it can prove fatal not only in the neonatal period but also during adult life. Here, we describe a case with a novel homozygous UGT1A1 p.Pro176Leu mutation. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
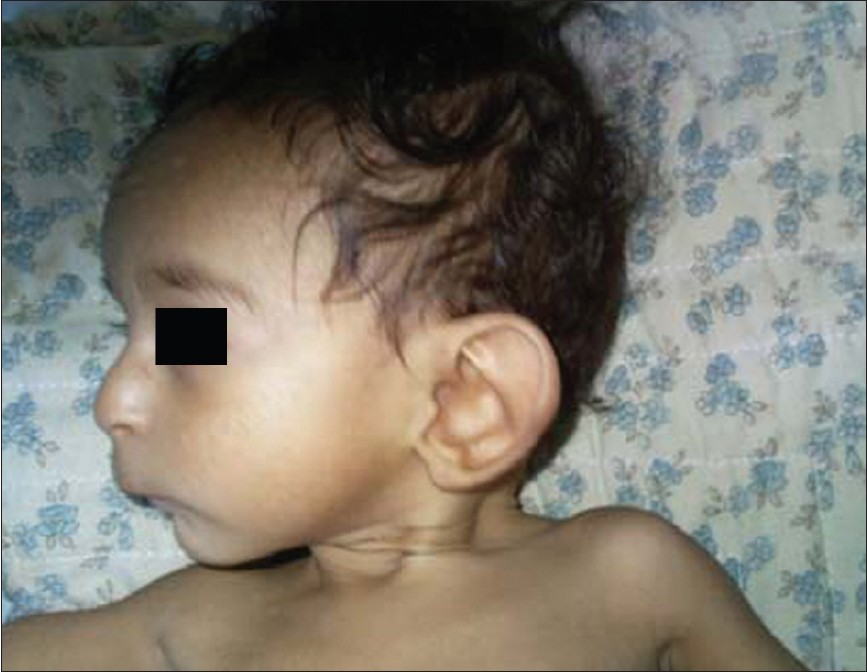 |
Smith-Lemli-Opitz-syndrome |
p. 235 |
Rachana Gedam, Ira Shah, Uma Ali, Alpana Ohri
DOI:10.4103/0971-6866.100779 PMID:23162303Smith-Lemli-Opitz syndrome is an autosomal recessively inherited disorder. A severe defect in cholesterol biosynthesis has been identified leading to abnormally low plasma cholesterol levels and elevated levels of the cholesterol precursor 7-dehydrocholesterol, the result of deficiency of 7-dehydrocholesterol reductase. We describe one such child with Smith-Lemli-Opitz syndrome. This child had clinical features similar to Smith-Lemli-Opitz syndrome like facial dysmorphism and cardiac and renal anomalies with failure to thrive. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
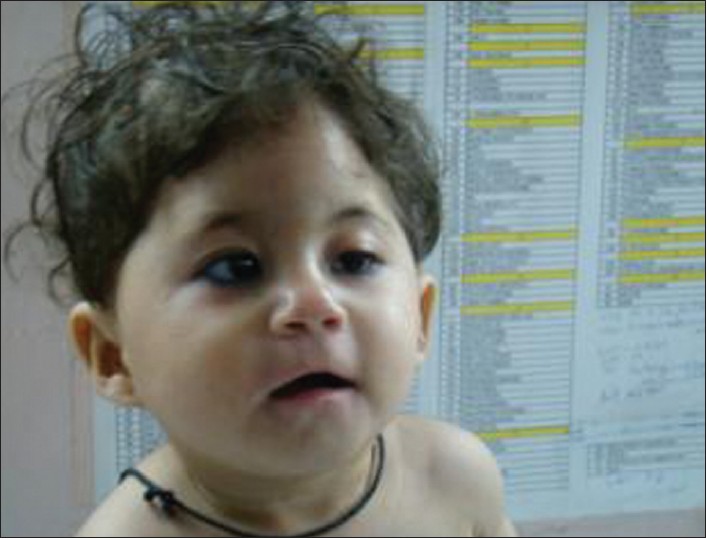 |
A familial deletion 4q syndrome: An outcome of a paracentric inversion |
p. 238 |
Meena Lall, Ratna Puri, Pushpa Saviour, Ishwar Verma
DOI:10.4103/0971-6866.100780 PMID:23162304Chromosome inversions are intra-chromosomal rearrangements formed when the chromosome breaks occur at two places, and in the process of repair the intervening segments are joined in an inverted or opposite manner. Inversions themselves do not appear to cause clinical anomalies, if balanced. Abnormal phenotypes can occur due to gene disruption at the point of breakage and reunion or due to duplication/deficiency recombinants formed during crossover at meiosis. We report a case with familial deletion 4q syndrome in a 1-year-old female child with dysmorphism and congenital abnormalities. The deletion was an outcome of a paracentric inversion 4q31.2q35.2. The deletion was confirmed by fluorescence in situ hybridization using telomeric DNA probes for chromosome No. 4. An attempt was made to correlate the genotype with the phenotype. The father had the same rearrangement with a milder phenotype. The recurrence risk in such cases is high. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
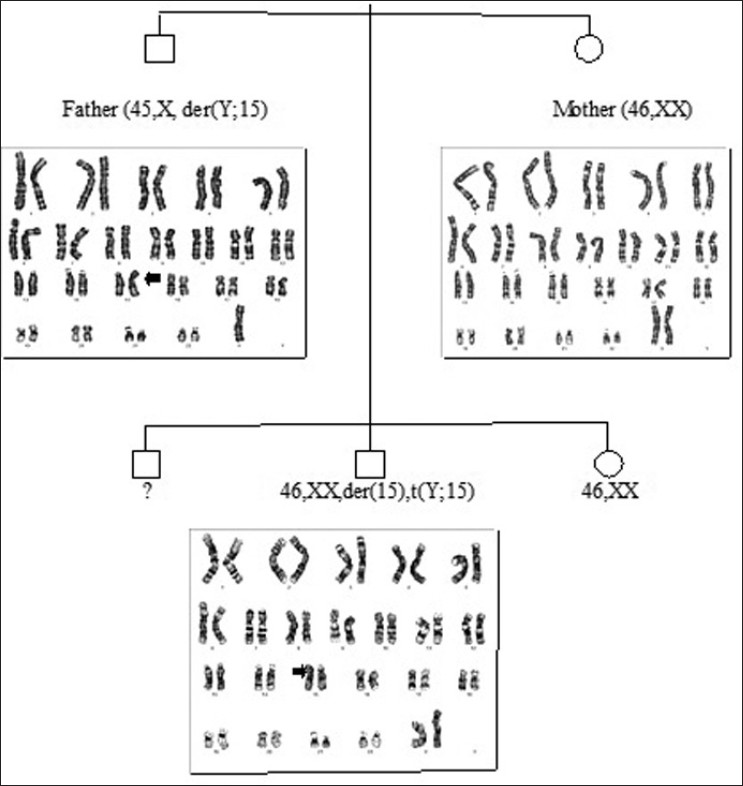 |
46,XX, der(15),t(Y;15)(q12;p11) karyotype in an azoospermic male  |
p. 241 |
Serap T Onrat, Zafer Söylemez, Muhsin Elmas
DOI:10.4103/0971-6866.100785 PMID:23162305We report on a Yq/15p translocation in a 23-year-old infertile male referred for Klinefelter Syndrome testing, who had azoospermia and bilateral small testes. Hormonal studies revealed hypergonadotropic hypogonadism. Conventional cytogenetic procedures giemsa trypsin giemsa (GTG) and high resolution banding (HRB) and molecular cytogenetic techniques Fluorescence In Situ Hybridization (FISH) performed on high-resolution lymphocyte chromosomes revealed the karyotype 46,XX, t(Y;15)(q12;p11). SRY-gene was confirmed to be present by classical Polymerase Chain Reaction (PCR) methods. His father carried de novo derivative chromosome 15 [45,X, t(Y;15)(q12;p11)] and was fertile; the karyotype of the father using G-band technique confirmed a reciprocal balanced translocation between chromosome Y and 15. In the proband, the der (15) has been inherited from the father because the mother had a normal karyotype (46,XX). In the proband, the der (15) could have produced genetic imbalance leading to unbalanced robertson translocation between chromosome Y and 15, which might have resulted in azoospermia and infertility in the proband. The paternal translocation might have lead to formation of imbalanced ova, which might be resulted infertility in the proband. Sister's karyotypes was normal (46,XX) while his brother was not analyzed. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
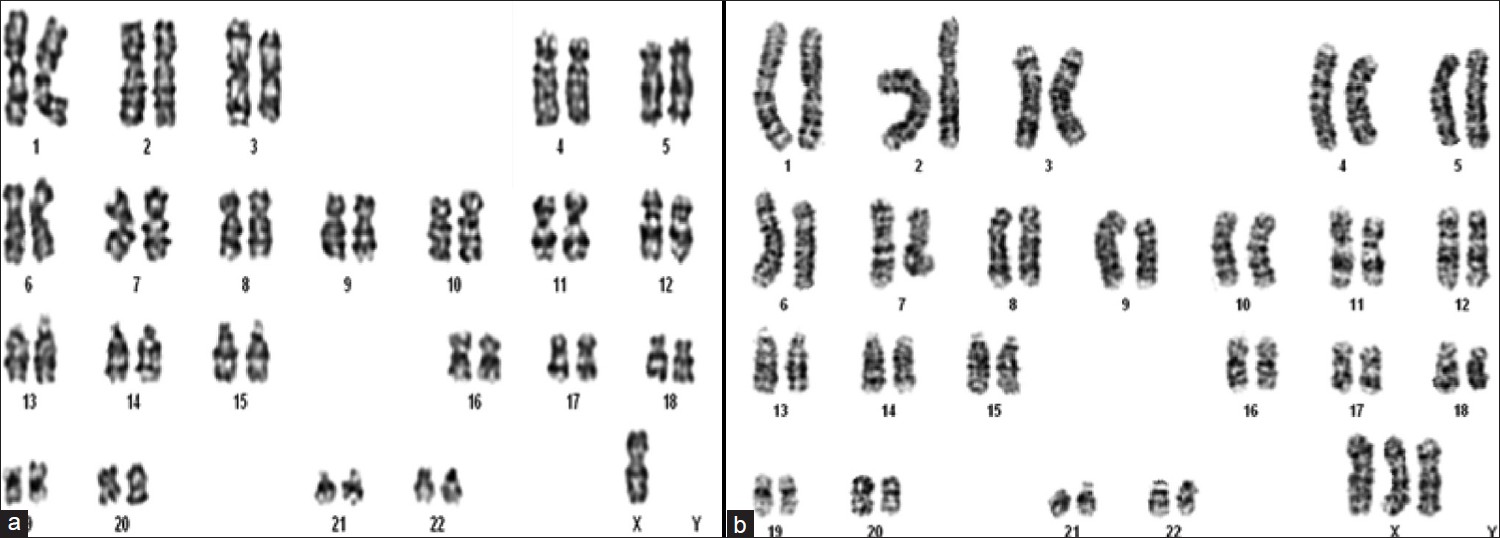 |
Mosaic triple X syndrome in a female with primary amenorrhea |
p. 246 |
A Venkateshwari, K Srimanjari, A Srilekha, Ashrafunnisa Begum, M Sujatha, T Sunitha, Pratibha Nallari, A Jyothy
DOI:10.4103/0971-6866.100790 PMID:23162306Background: Turner's syndrome is the most common chromosomal abnormality in females, affecting 1 in 2,500 live female births. It is a result of absence of an X chromosome or the presence of a structurally abnormal X chromosome. Its most consistent clinical features are short stature and ovarian failure.
Aim: The aim of the study was to report a rare case of mosaic triple X syndrome in a female with primary amenorrhea.
Materials and Methods: The chromosomal analysis using GTG banding was carried out, which revealed a mosaicism with 45,XO/47,XXX chromosomal constitution. Fluorescent in situ hybridization was also carried out to further confirm the observation made in the study.
Conclusion: The physical features presented by the female could be due to the 45,XO/47,XXX mosaicism and the karyotype analysis was consistent with the diagnosis and clinical symptoms. Triple X mosaicism was confirmed with conventional and molecular cytogenetic analysis. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
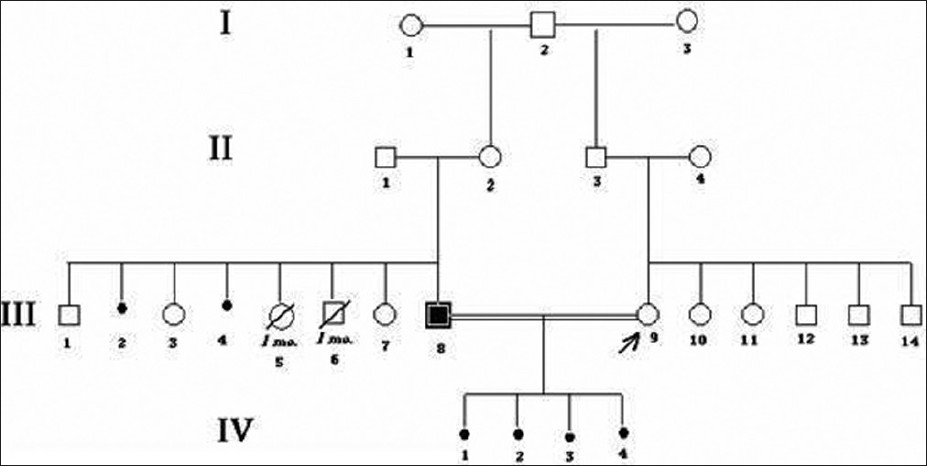 |
Genetic counseling in carriers of reciprocal translocations involving two autosomes |
p. 250 |
Bahareh Pourjafari, Hamid Pour-Jafari, Marzieh Farimani, Safieh Ghahramani, Ebrahim Kamrani Saleh
DOI:10.4103/0971-6866.100802 PMID:23162307One of the main genetic causes involve in the pathogenesis of recurrent abortion is parental chromosomal abnormalities. The central concept in genetic counseling with such families is to estimate the probability of recurrence of unfavorable pregnancy outcomes. The main questions that consultants usually ask are: Why did this happen? What is the risk to be done again?
Our cases were two families with repeated miscarriage. The pedigrees were drawn, the chromosomes of couples were studied, and estimation for recurrent risk was done. We tried to answer those two main questions and clear the results for them.
Parental chromosome abnormalities were founded after karyotyping with GTG technique at 450 band resolution, revealing 46 chromosomes with balanced translocation of autosomes in one of the partner in both families. Recurrent risk was estimated as "high" for their future pregnancies in each family.
Couples in which one partner is the carrier of such balanced translocation have increased risks of infertility, recurrent abortion, and delivery of chromosomally abnormal offspring. Genetic counseling of such couples, therefore, presents a unique challenge and should be considered in dealing with such families. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Waardenburg syndrome: A rare genetic disorder, a report of two cases |
p. 254 |
Sudesh Kumar, Kiran Rao
DOI:10.4103/0971-6866.100804 PMID:23162308Waardenburg syndrome (WS) is a rare genetic disorder. Patients have heterochromia or eyes with iris of different color, increased inter-canthal distance, distopia canthorum, pigmentation anomalies, and varying degree of deafness. It usually follows autosomal dominant pattern. In this report, two cases have been discussed but no familial history of WS has been found. Counseling of the patient is necessary and cases of irreversible deafness have been treated. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Unusual manifestation of Marden-walker syndrome |
p. 256 |
Amar M Taksande, KY Vilhekar
DOI:10.4103/0971-6866.100798 PMID:23162309Marden-Walker syndrome (MWS) is characterized by multiple joint contractures, a mask-like face with blepharophimosis, micrognathia, high-arched or cleft palate, low-set ears, decreased muscular bulk, arachnodactyly, and kyphoscoliosis. We report a case of MWS along with unusual manifestation of neurological, cardiovascular, and genitourinary system. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
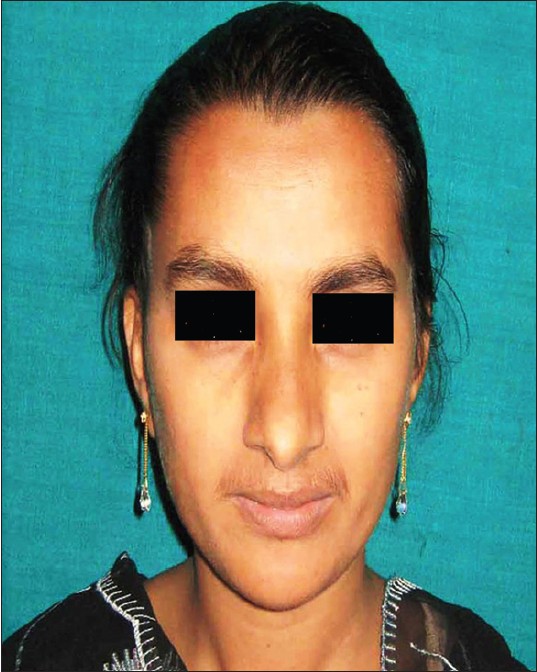 |
Association of generalized aggressive periodontitis and ectrodactyly-ectodermal dysplasia-cleft syndrome |
p. 259 |
Rosamma Joseph, Sameera G Nath
DOI:10.4103/0971-6866.100793 PMID:23162310Ectrodactyly-ectodermal dysplasia-cleft (EEC) syndrome is an autosomal dominant disorder characterized by the triad of ectrodactyly, ectodermal dysplasia, and facial clefting. Even though literature has documented the association of various genetic disorders with aggressive periodontitis, the periodontal manifestations in patients with EEC syndrome have never been addressed. This case report presents the periodontal status of three patients in a family with EEC syndrome. The presence of generalized aggressive periodontitis was noticed in these patients. EEC syndrome could be a new addition to the group of genetic disorders associated with aggressive periodontitis. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
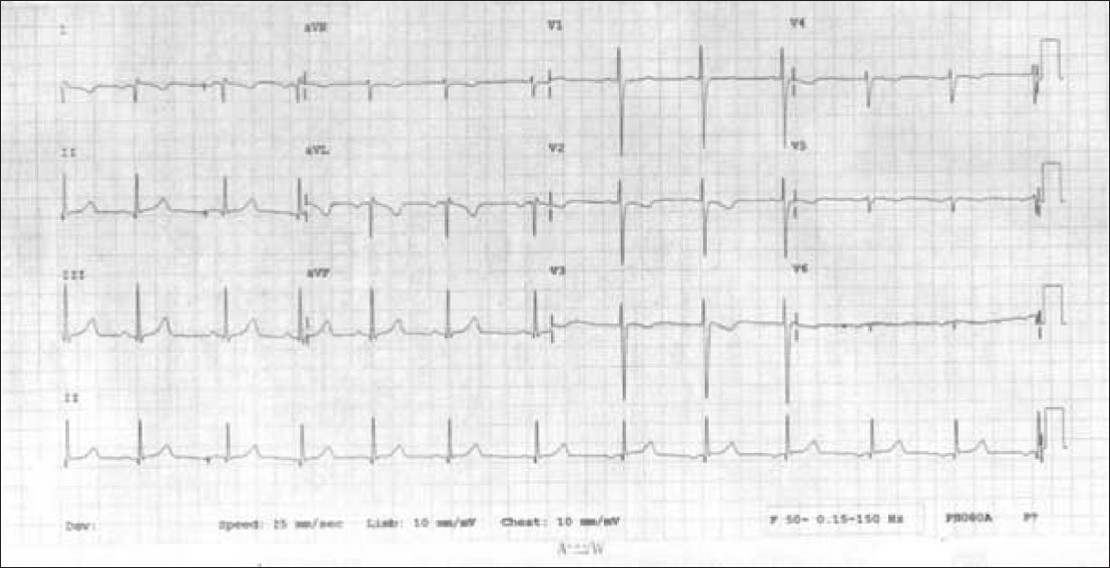 |
A case of Kartagener's syndrome: Importance of early diagnosis and treatment |
p. 263 |
Sanjay Gupta, Kumud K Handa, Ravi R Kasliwal, Pankaj Bajpai
DOI:10.4103/0971-6866.100787 PMID:23162311Kartagener's syndrome is a very rare congenital malformation comprising of a classic triad of sinusitis, situs inversus and bronchiectasis. Primary ciliary dyskinesia is a genetic disorder with manifestations present from early life and this distinguishes it from acquired mucociliary disorders. Approximately one half of patients with primary ciliary dyskinesia have situs inversus and, thus are having Kartagener syndrome. We present a case of 12 year old boy with sinusitis, situs inversus and bronchiectasis. The correct diagnosis of this rare congenital autosomal recessive disorder in early life is important in the overall prognosis of the syndrome, as many of the complications can be prevented if timely management is instituted, as was done in this in this case. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| LETTERS TO THE EDITOR |
|
|
|
|
An unusual clonal cytogenetic abnormality with t(15;17)(p11;q21) in a patient with severe aplastic anemia |
p. 268 |
Sanjeev K Sharma, Narendra Agrawal, Sonal Jain, Mohit Chowdhry, Pawan K Singh, Tulika Seth, Pravas Mishra, Manoranjan Mahapatra, Seema Tyagi, Haraprasad Pati
DOI:10.4103/0971-6866.100782 PMID:23162312 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Honey, chromosomal breakage and fanconi anemia |
p. 269 |
Somsri Wiwanitkit, Viroj Wiwanitkit
DOI:10.4103/0971-6866.100783 PMID:23162313 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Large scale meta-analysis of genetic studies in ischemic stroke: Five genes involving 152,297 individuals |
p. 270 |
Amit Kumar, Pradeep Kumar, Jitendra K Sahu
DOI:10.4103/0971-6866.100786 PMID:23162314 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|