|
Show all abstracts Show selected abstracts Add to my list |
|
| EDITORIAL |
|
|
|
Prenatal diagnosis of common fetal aneuploidies: Scenario in India |
p. 1 |
V Baburao, Ajit C Gorakshakar
DOI:10.4103/0971-6866.112868 PMID:23901185 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| REVIEW ARTICLE |
 |
|
|
|
Delivery systems for gene therapy |
p. 3 |
Shrikant Mali
DOI:10.4103/0971-6866.112870 PMID:23901186The structure of DNA was unraveled by Watson and Crick in 1953, and two decades later Arber, Nathans and Smith discovered DNA restriction enzymes, which led to the rapid growth in the field of recombinant DNA technology. From expressing cloned genes in bacteria to expressing foreign DNA in transgenic animals, DNA is now slated to be used as a therapeutic agent to replace defective genes in patients suffering from genetic disorders or to kill tumor cells in cancer patients. Gene therapy provides modern medicine with new perspectives that were unthinkable two decades ago. Progress in molecular biology and especially, molecular medicine is now changing the basics of clinical medicine. A variety of viral and non-viral possibilities are available for basic and clinical research. This review summarizes the delivery routes and methods for gene transfer used in gene therapy. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| ORIGINAL ARTICLES |
|
|
|
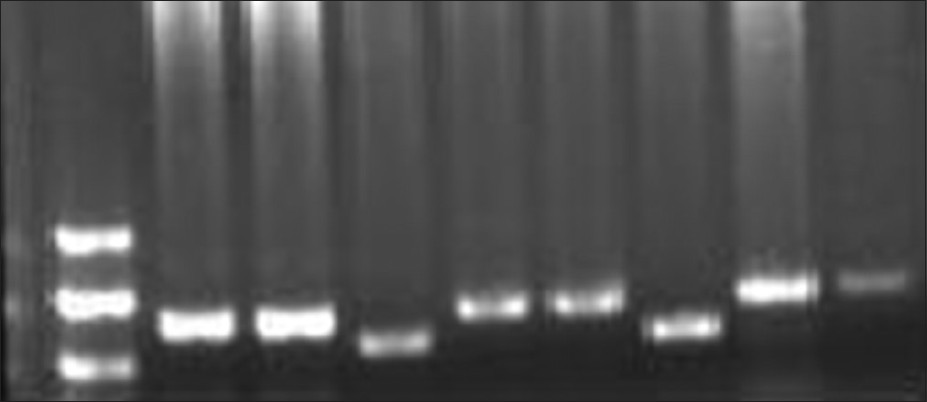  |
Nucleotide sequence analysis of NIPBL gene in Indian Cornelia de Lange syndrome cases |
p. 9 |
Shailesh Bajaj, Suvidya Ranade, Prakash Gambhir
PMID:23901187Background: Cornelia de Lange syndrome (CdLS) is a multisystem developmental disorder in children. The disorder is caused mainly due to mutations in Nipped-B-like protein. The molecular data for CdLS is available from developed countries, but not available in developing countries like India. In the present study, the hotspot region of NIPBL gene was screened by Polymerase Chain Reaction which includes exon 2, 22, 42, and a biggest exon 10, in six CdLS patients and ten controls.
Materials and Methods: The method adopted in present study was amplification of the target exon by using polymerase chain reaction, qualitative confirmation of amplicons by Agarose Gel Electrophoresis and use of amplicons for Conformation Sensitive Gel Electrophoresis to find heteroduplex formation followed by sequencing.
Results: We report two polymorphisms in the studied region of gene NIPBL. The polymorphisms are in the region of intron 1 and in exon 10. The polymorphism C/A is present in intron 1 region and polymorphism T/G in exon 10.
Conclusion: The intronic region polymorphism may have a role in intron splicing whereas the polymorphism in exon 10 results in amino acid change (Val to Gly). These polymorphisms are disease associated as these are found in CdLS patients only and not in controls. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
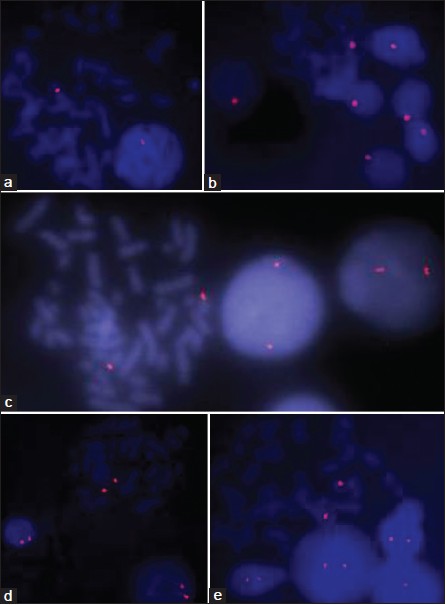 |
Rapid detection of chromosome X, Y, 13, 18, and 21 aneuploidies by primed in situ labeling/synthesis technique |
p. 14 |
Ashutosh Halder, Manish Jain, Isha Chaudhary
DOI:10.4103/0971-6866.112877 PMID:23901188Aims and Objective: Primed in situ labeling/synthesis (PRINS) technique is an alternative to fluorescent in situ hybridization for chromosome analysis. This study was designed to evaluate the application of PRINS for rapid diagnosis of common chromosomal aneuploidy.
Materials and Methods : We have carried out PRINS using centromere specific oligonucleotide primers for chromosome X, Y, 13, 18 and 21 on lymphocyte metaphase and interphase cells spread. Specific primer was annealed in situ, followed by elongation of primer by Taq DNA polymerase in presence of labeled nucleotides. Finally, reaction was stopped and visualized directly under fluorescent microscope.
Results: Discrete centromere specific signals were observed with each primer.
Conclusion: PRINS seems to be a rapid and reliable method to detect common chromosome aneuploidy in peripheral blood lymphocyte metaphase and interphase cells. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
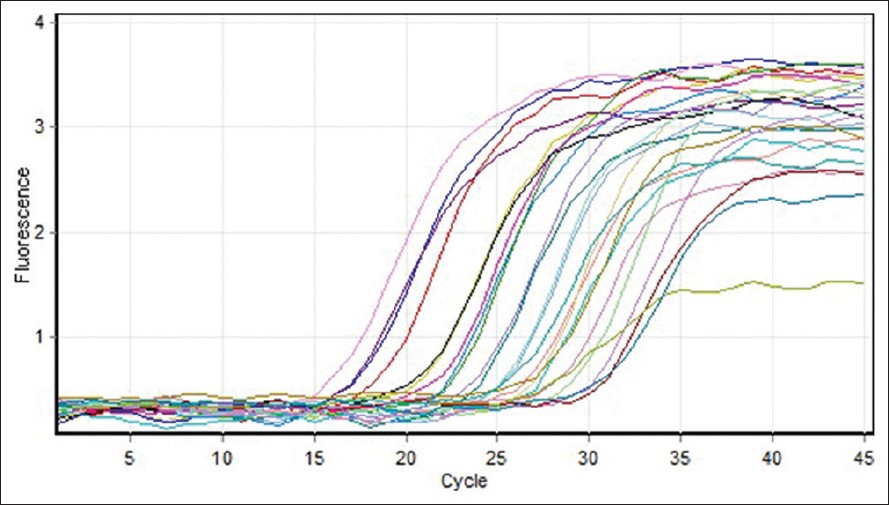 |
Investigation of cytocrom c oxidase gene subunits expression on the Multiple sclerosis |
p. 18 |
Naeimeh Safavizadeh, Seyed Ali Rahmani, Mohamad Zaefizadeh
DOI:10.4103/0971-6866.112879 PMID:23901189Introduction: Multiple sclerosis (MS) is an autoimmune inflamatory disease, which affects the (Central Nervous System) and leads to the destruction of myelin and atrophy of the axons. Genetic factors, in addition to environmental ones, seem to play a role in MS. Numerous studies have reported mitochondrial defects including a reduction in cytochrome c oxidase (COX) complex function related to the reduction of mitochondrial genes expression in the cortex tissue of patients with MS have been reported.
Materials and Methods: This study aimed to assess COX5B and COX2 genes expression in MS patients and compare it with normal subjects. We determine expression levels of genes COX5B and COX2, and also gene reference ß-actine using real-time polymerase chain reaction (RT-PCR) method. Data were obtained and obtained and standardized with the gene reference and were analyzed using independent sample t-test with SPSS and Excel programs.
Result and Discussion: The resultshowed COX5B gene expression reduced signifi cant in MS patients compared to normal subjects (P < -0.05) whereas, there was no significant difference in the COX2 gene expression between normal subjects and patients. Thus, it can be claimed that down-regulation of mitochondrial electron transport chain genes supported the hypothesis that hypoxia-like tissue injury in MS may be due to mitochondrial genes, different expression impairment. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Association of apolipoprotein B XbaI gene polymorphism and lipid profile in northern Indian obese |
p. 26 |
Neena Srivastava, Jai Prakash, Apurva Srivastava, Chandra Gupta Agarwal, Deep Chandra Pant, Balraj Mittal
DOI:10.4103/0971-6866.112880 PMID:23901190Background: Over the last few decades, obesity, diabetes, and hypertension have become main health evils. The health problems of obesity are well-recognized. However, the fact that all obese individuals are not at the same risk of developing a disease is also recognized. The apolipoprotein B (APOB) plays a central role in lipid metabolism. So we compare the association of APOB XbaI gene polymorphism and lipid profile total in obese north Indian population.
Materials and Methods: A total of 132 obese (body mass index [BMI] >25 kg/m 2 ) and 132 age matched non-obese (BMI ≤ 25 kg/m 2 ) subjects were studied after taking detailed clinical profile. Lipid profile in serum/plasma was done using commercial kits. Genetic analysis of APOB XbaI was done using Polymerase Chain Reaction-Restriction Fragment Leanth polymorphism (PCR-RFLP).
Statistical Analysis: Statistical analysis was performed by Statistical Package for the Social Sciences (SPSS) (version 11.5) software (IBM Corporation). All continuous variables were expressed as mean ± SD and tested by analysis of variance test. Comparisons of categorical variables were assessed using χ2 tests or Fisher's exact test. P < 0.05 was considered as significant.
Results: Analysis showed that obese subjects had significantly higher value of the waist-to-hip ratio, blood pressure (systolic and diastolic), and lipid profile. In APOB XbaI gene polymorphism, we did not find significant differences in genotype or allele frequencies. Moreover, none of the studied metabolic parameters (lipid profile) showed any association with the gene polymorphism.
Conclusions: Study reveals no considerable association of APOB XbaI gene polymorphism with obesity and lipid profile in north Indians. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
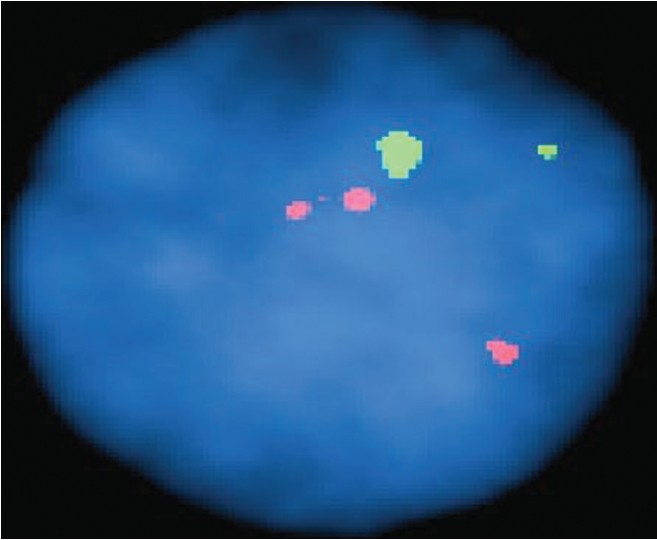 |
Rapid-prenatal diagnosis through fluorescence in situ hybridization for preventing aneuploidy related birth defects |
p. 32 |
Ashish Fauzdar, Mohit Chowdhry, RN Makroo, Manoj Mishra, Priyanka Srivastava, Richa Tyagi, Preeti Bhadauria, Anita Kaul
DOI:10.4103/0971-6866.112881 PMID:23901191Background and Objective: Women with high-risk pregnancies are offered prenatal diagnosis through amniocentesis for cytogenetic analysis of fetal cells. The aim of this study was to evaluate the effectiveness of the rapid fluorescence in situ hybridization (FISH) technique for detecting numerical aberrations of chromosomes 13, 21, 18, X and Y in high-risk pregnancies in an Indian scenario.
Materials and Methods: A total of 163 samples were received for a FISH and/or a full karyotype for prenatal diagnosis from high-risk pregnancies. In 116 samples both conventional culture techniques for getting karyotype through G-banding techniques were applied in conjunction to FISH test using the AneuVysion kit (Abbott Molecular, Inc.), following standard recommended protocol to compare the both the techniques in our setup.
Results: Out of 116 patients, we got 96 normal for the five major chromosome abnormality and seven patients were found to be abnormal (04 trisomy 21, 02 monosomy X, and 01 trisomy 13) and all the FISH results correlated with conventional cytogenetics. To summarize the results of total 163 patients for the major chromosomal abnormalities analyzed by both/or cytogenetics and FISH there were 140 (86%) normal, 9 (6%) cases were abnormal and another 4 (2.5%) cases were suspicious mosaic and 10 (6%) cases of culture failure. The diagnostic detection rate with FISH in 116 patients was 97.5%. There were no false-positive and false-negative autosomal or sex chromosomal results, within our established criteria for reporting FISH signals.
Conclusion: Rapid FISH is a reliable and prompt method for detecting numerical chromosomal aberrations and has now been implemented as a routine diagnostic procedure for detection of fetal aneuploidy in India. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
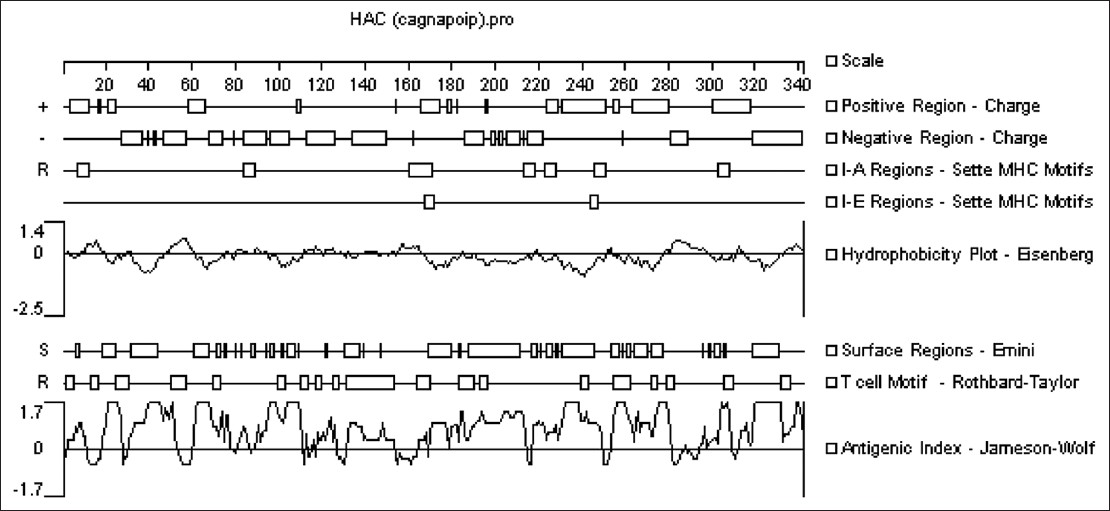 |
In silico experiment with an-antigen-toll like receptor-5 agonist fusion construct for immunogenic application to Helicobacter pylori |
p. 43 |
Mohamad Ali Haghighi, Ashraf Mohabati Mobarez, Ali Hatef Salmanian, Mohamad Moazeni, Mohamad Reza Zali, Mehdi Sadeghi, Jafar Amani
DOI:10.4103/0971-6866.112885 PMID:23901192Backgrounds: Helicobacter pylori colonize the gastric mucosa of half of the world's population. Although it is classified as a definitive type I carcinogen by World Health Organization, there is no effective vaccine against this bacterium. H. pylori evade the host immune response by avoiding toll-like detection, such as detection via toll-like receptor-5 (TLR-5). Thus, a chimeric construct consisting of selected epitopes from virulence factors that is incorporated into a TLR-5 ligand (Pseudomonas flagellin) could result in more potent innate and adaptive immune responses.
Materials and Methods: Based on the histocompatibility antigens of BALB/c mice, in silico techniques were used to select several fragments from H. pylori virulence factors with a high density of B- and T-cell epitopes.
Results: These segments consist of cytotoxin-associated geneA (residue 162-283), neutrophil activating protein (residue 30-135) and outer inflammatory protein A (residue 155-268). The secondary and tertiary structure of the chimeric constructs and other bioinformatics analyses such as stability, solubility, and antigenicity were performed. The chimeric construct containing antigenic segments of H. pylori proteins was fused with the D3 domain of Pseudomonas flagellin. This recombinant chimeric gene was optimized for expression in Escherichia coli. The in silico results showed that the conserved C- and N-terminal domains of flagellin and the antigenicity of selected fragments were retained.
Discussion: In silico analysis showed that Pseudomonas flagellin is a suitable platform for incorporation of an antigenic construct from H. pylori. This strategy may be an effective tool for the control of H. pylori and other persistent infections. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
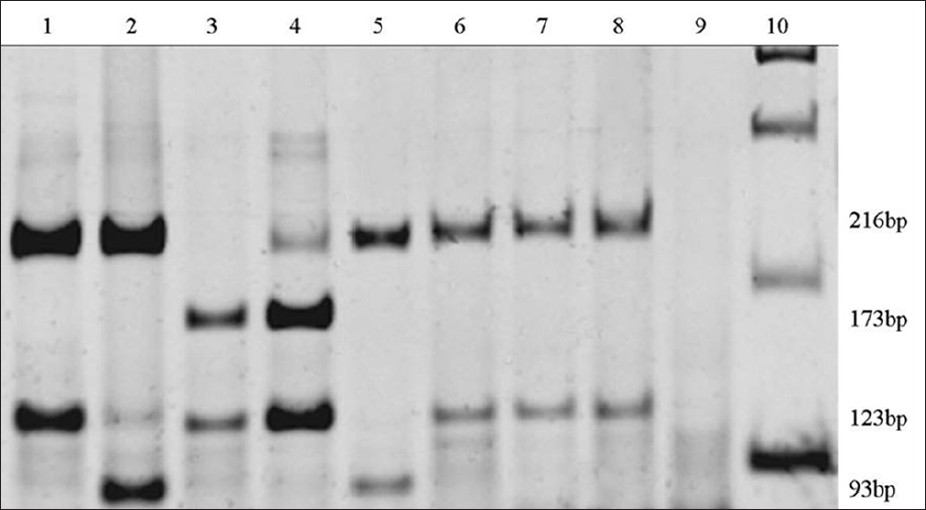 |
Investigation of the A1555G mutation in mitochondrial DNA (MT-RNR1) in groups of Brazilian individuals with nonsyndromic deafness and normal-hearing |
p. 54 |
Karina Bezerra Salomão, Christiane Maria Ayo, Valter Augusto Della-Rosa
DOI:10.4103/0971-6866.112888 PMID:23901193Background: Mutations of mitochondrial DNA were described into two genes: The mitochondrially encoded 12S RNA (MT-RNR1) and the mitochondrially encoded tRNA serine ucn (MT-TS1). The A1555G mutation in MT-RNR1 gene is a frequent cause of deafness in different countries.
Aim: The aim of this work was to investigate the frequency of the A1555G mutation in the MT-RNR1 gene in the mitochondrial DNA in Brazilians individuals with nonsyndromic deafness, and listeners.
Materials and Methods: DNA samples were submitted to polymerase chain reaction and to posterior digestion with the Hae III enzyme.
Results: Seventy eight (78) DNA samples of deaf individuals were analyzed; 75 showed normality in the region investigated, two samples (2.5%) showed the T1291C substitution, which is not related to the cause of deafness, and one sample (1.3%) showed the A1555G mutation. Among the 70 non-impaired individuals no A1555G mutation or T1291C substitution was found.
Conclusion: We can affirm that A1555G mutation is not prevalent, or it must be very rare in normal-hearing subjects in the State of Paranα, the south region of Brazil. The A1555G mutation frequency (1.3%) found in individual with nonsyndromic deafness is similar to those found in other populations, with nonsyndromic deafness. Consequently, it should be examined in deafness diagnosis. The investigation of the A1555G mutation can contribute towards the determination of the nonsyndromic deafness etiology, hence, contributing to the correct genetic counseling process. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Association of interleukin-4 and interleukin-17F polymorphisms in periodontitis in Dravidian ethnicity |
p. 58 |
Nidhi Jain, Rosamma Joseph, Shabeesh Balan, R Arun, Moinak Banerjee
DOI:10.4103/0971-6866.112891 PMID:23901194Background: Complex network of pro and anti-inflammatory cytokines are known to act in inflamed periodontal tissue. This study explores the distribution of interleukin (IL)-4 (+33 C/T) and IL-17F (7383A/G, 7488A/G) gene polymorphism in chronic and aggressive periodontitis subjects of Dravidian ethnicity.
Materials and Methods: This case control study consisted of 124 periodontitis individuals comprising of 63 chronic and 61 aggressive periodontitis subjects as cases, and control group consisted of 101 healthy subjects. All subjects were genotyped for IL-4 + 33C/T, IL-17F 7383A/G, 7488A/G by polymerase chain reaction amplification followed by TaqMan assay for IL-4 + 33C/T, restriction enzyme digestion and gel electrophoresis for IL-17F 7383A/G and sequencing for IL-17F 7488A/G.
Results: IL-4 + 33C/T was significantly associated with periodontitis (P < 0.05) at both allelic and genotypic level. In subgroup analysis also significant difference (P < 0.05) in allelic distribution between aggressive periodontitis and control group for loci IL-4 + 33C/T was noted. However, there was a lack of association between IL-17F 7383A/G and IL-17F 7488A/G with periodontitis and its sub-groups at both allelic and genotypic levels.
Conclusions: In Malayalam speaking Dravidian population IL-4 + 33C/T loci appears to be an important risk factor for periodontal disease with a leaning towards aggressive periodontitis. The association between IL-17F at 7383A/G and 7488A/G loci with either chronic or an aggressive periodontitis could not be ascertained. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
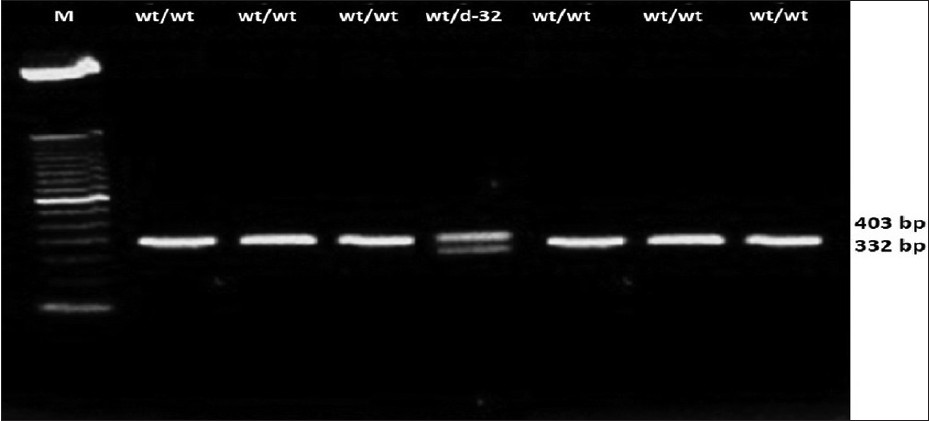 |
Distribution of CC-chemokine receptor-5-∆32 allele among the tribal and caste population of Vidarbha region of Maharashtra state |
p. 65 |
Arvind B Chavhan, Santosh S Pawar, Rajusing G Jadhao, Kishor G Patil
DOI:10.4103/0971-6866.112894 PMID:23901195Background: Genetic relationships among the ethnic groups are not uniform across the geographical region. Considering this assumption, we analyzed the frequency of the CC-chemokine receptor-5 (CCR5)-∆32 allele of the CCR5 chemokine receptor, which is considered a Caucasian marker, in Bhil tribal and Brahmin caste sample sets from the population.
Materials and Methods: 108 blood samples were collected from 6 tribe's populations and a caste population from the district of Vidarbha region.
Results and Discussion: The presence of low frequencies of CCR5-Δ32 in an individual of Bhil tribe (0.034, χ2 value 0.017) in the present study implies that these communities may have a better resistance toward human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS) than the other studied tribe sample, as non-show such mutation.
Conclusion: The marginal presence of the allele seen in the studied tribal population could be due to gene flow from the people of European descent. However, lack of the homozygous CCR5-Δ32 mutation and the low prevalence of heterozygous CCR5-Δ32 mutations suggest that the Indians are highly susceptible to HIV/AIDS, and this correlates with the highest number of HIV/AIDS infected individuals in India. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
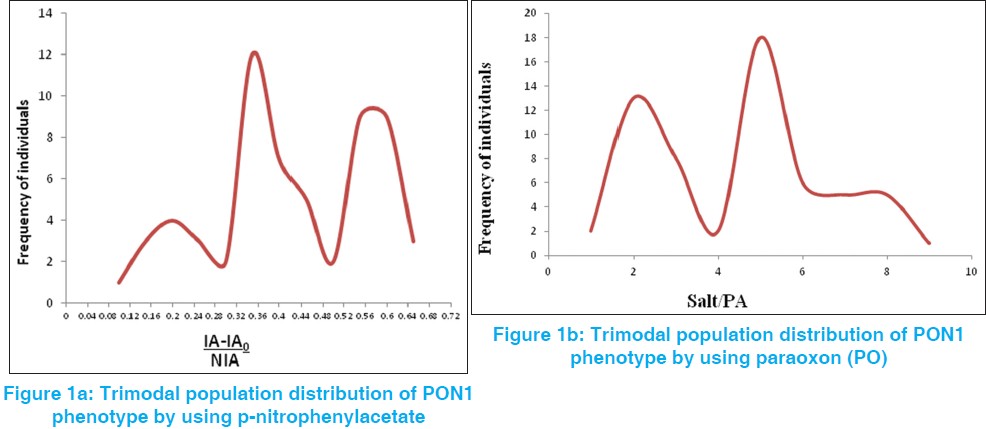 |
The determination of Q192R polymorphism of paraoxonase 1 by using non-toxic substrate p-nitrophenylacetate |
p. 71 |
MR Mogarekar, Seema S Chawhan
DOI:10.4103/0971-6866.112897 PMID:23901196Context: The human serum paraoxonase 1 (PON1) is calcium-dependent esterase and associates with the high density serum lipoproteins. PON1 plays a major role in oxidation of high density lipoprotein and low density lipoprotein and prevention of atherogenesis in coronary heart disease. PON1Q and R allele hydrolyses number of substrates like paraoxon (PO) (diethyl p-nitrophenyl phosphate) and phenylacetate.
Aims: The aim of the study is to the determination of Q192R polymorphism of PON1 by using non-toxic substrate p-nitrophenylacetate and compares it with the phenotype determined by using PO as substrate.
Materials and Methods: The study group consists of 60 healthy normal patients. Paraoxonase activity was measured using the procedure described by Eckerson (Reference method) and for phenotyping; the ratio of hydrolysis of PO in the presence of 1 M NaCl (salt-stimulated PON1, SALT) to the hydrolysis of phenylacetate (PA) is calculated. In new method (Haagen et al.) arylesterase activity measured using p-nitrophenylacetate and for phenotyping arylesterase, the ratio of inhibition of enzymatic hydrolysis of p-nitrophenylacetate (substrate) by phenyl acetate to non-inhibited hydrolysis of p-nitrophenylacetate (inhibited arylesterase activity (IA-IA 0 )/non-inhibited arylesterase activity (NIA).
Results: It was found that paraoxonase activity is trimodally distributed in both the methods. There is no significant difference in the distribution of PON1 phenotypes of both reference method and new method being frequencies 0.946 and 0.376 respectively and there was no significant difference for phenotypic polymorphism for an individual by both methods (χ2 = 0.15 and P = 0.9262).
Conclusion: The Q192R polymorphism of PON1 by using non-toxic substrate p-nitrophenylacetate showed trimodal distribution of QQ (homozygous), QR (heterozygous), and RR (homozygous) phenotype and it is comparable with reference method. This method can be used for PON1 phenotype in different pathological and complex disease conditions. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| BRIEF REPORT |
|
|
|
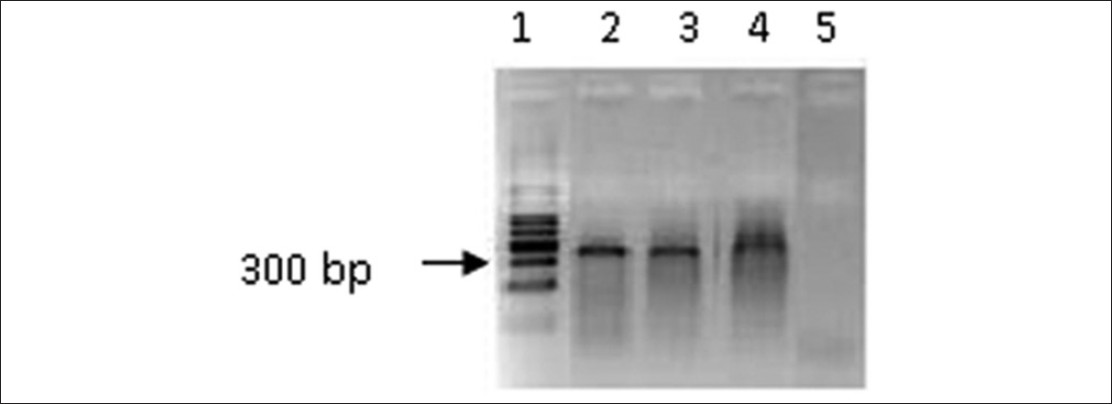 |
Polymerase chain reaction optimization for amplification of Guanine-Cytosine rich templates using buccal cell DNA |
p. 78 |
C. H. W. M. R. Chandrasekara Bhagya, WS Wijesundera Sulochana, N Perera Hemamali
DOI:10.4103/0971-6866.112898 PMID:23901197Context: Amplification of Guanine-Cytosine (GC) -rich sequences becomes important in screening and diagnosis of certain genetic diseases such as diseases arising due to expansion of GC-rich trinucleotide repeat regions. However, GC-rich sequences in the genome are refractory to standard polymerase chain reaction (PCR) amplification and require a special reaction conditions and/or modified PCR cycle parameters.
Aim: Optimize a cost effective PCR assay to amplify the GC-rich DNA templates.
Settings and Design: Fragile X mental retardation gene (FMR 1) is an ideal candidate for PCR optimization as its GC content is more than 80%. Primers designed to amplify the GC rich 5' untranslated region of the FMR 1 gene, was selected for the optimization of amplification using DNA extracted from buccal mucosal cells.
Materials and Methods: A simple and rapid protocol was used to extract DNA from buccal cells. PCR optimization was carried out using three methods, (a) substituting a substrate analog 7-deaza-dGTP to dGTP (b) in the presence of a single PCR additive and (c) using a combination of PCR additives. All PCR amplifications were carried out using a low-cost thermostable polymerase.
Results: Optimum PCR conditions were achieved when a combination of 1M betaine and 5% dimethyl sulfoxide (DMSO) was used.
Conclusions: It was possible to amplify the GC rich region of FMR 1 gene with reproducibility in the presence of betaine and DMSO as additives without the use of commercially available kits for DNA extraction and the expensive thermostable polymerases. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| CASE REPORTS |
|
|
|
|
Fanconi-Bickel syndrome versus osteogenesis imperfeeta: An Iranian case with a novel mutation in glucose transporter 2 gene, and review of literature |
p. 84 |
Fatemeh Hadipour, Peymaneh Sarkheil, Mehrdad Noruzinia, Zahra Hadipour, Taghi Baghdadi, Yousef Shafeghati
DOI:10.4103/0971-6866.112906 PMID:23901198Fanconi-Bickel syndrome is an extremely rare hereditary metabolic disease, characterized by hepatomegaly due to glycogen storage, refractory hypophosphatemic rickets, marked growth retardation and proximal renal tubular acidosis. Recurrent bone fractures are one of the hallmark findings. It is a single gene disorder; the responsible gene belongs to the facilitative glucose transporters 2 (GLUT2) family gene or (SLC2A2) mapped to the q26.1-26.3 locus on chromosome 3, and encodes the GLUT protein 2. This protein is expressed in pancreatic ί-cells, hepatocytes, renal tubules, and intestinal mucosa. Several mutations in the GLUT2 gene have been reported in different ethnicities. Herein we report an Iranian girl with a missed diagnosis of osteogenesis imperfecta. She was referred with the history of frequent fractures, and severe motor delay and was suspected to osteogenesis imperfecta. Following the case we detected refractory rickets instead of OI, sever growth failure, proximal renal tubulopathy and RTA, and enlarged kidneys, progressive hepatomegaly, and GSD on liver biopsy. Glucose and galactose tolerance tests confirmed abnormal carbohydrate metabolism. Molecular analysis on GLUT2 gene revealed a homozygous novel mutation in exon 5; it was 15 nucleotide deletion and 7 nucleotide insertion and caused a frame shift mutation, produced a premature truncated protein (P.A229QFsX19). This mutation has not been reported before in the relevant literature. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
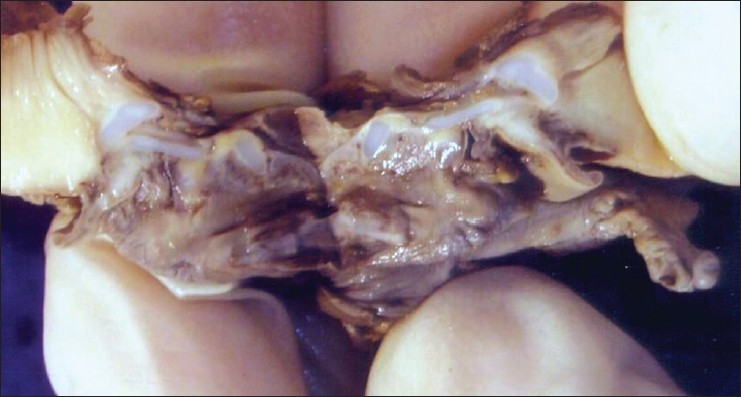 |
Tracheal agenesis with broncho-esophageal fistula in VACTERL / TACRD association |
p. 87 |
Suresh R. S. Mandrekar, Sangeeta Amoncar, R. G. W Pinto
DOI:10.4103/0971-6866.112910 PMID:23901199Tracheal agenesis (TA) is an extremely rare malformation. We report here autopsy findings in a case of TA with bronchoesophageal fistula of Floyd type III. The other malformations present included laryngeal atresia, Right lung hypolobulation, ventricular septal defect in membranous portion, bilateral cystic renal dysplasia, spleninculus, Meckel's diverticulum, and imperforate anus. The constellations of malformations present in our case have overlapping features with Vertebral anomalies, Anal atresia, Cardiovascular anomalies, Tracheo-esophageal fistula, Esophageal atresia, Renal anomalies, Limb anomalies and Tracheal atresia or laryngo tracheal atresia, Cardiac anomalies, Renal anomalies, Duodenal atresia association described previously in the literature. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Malignant infantile osteopetrosis |
p. 90 |
Kalenahalli Jagadish Kumar, Kasi Bandaru, Sathya Narayana Prashanth, Sangaraju Mamatha
DOI:10.4103/0971-6866.112911 PMID:23901200Osteopetrosis, a rare congenital genetic disease characterized by increased bone density due to impaired bone resorption by osteoclasts.It is classified into three forms: Infantile malignant autosomal recessive (AR) osteopetrosis, intermediate (AR) osteopetrosis and autosomal dominant (AD) osteopetrosis. Incidence of infantile malignant AR is 1/2,00,000 and if untreated has a fatal outcome.The condition is commonly diagnosed in infancy with symptoms of significant hematologic abnormalities with bone marrow failure, hepatosplenomegaly, macrocephaly with frontal bossing and bone fractures. Because of rarity of this type of malignant infantile form of osteopretrosis, we like to report this case of malignant infantile osteopetrosis who presented with bronchopneumonia, anemia with melaena at 2 months 15 days of age. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
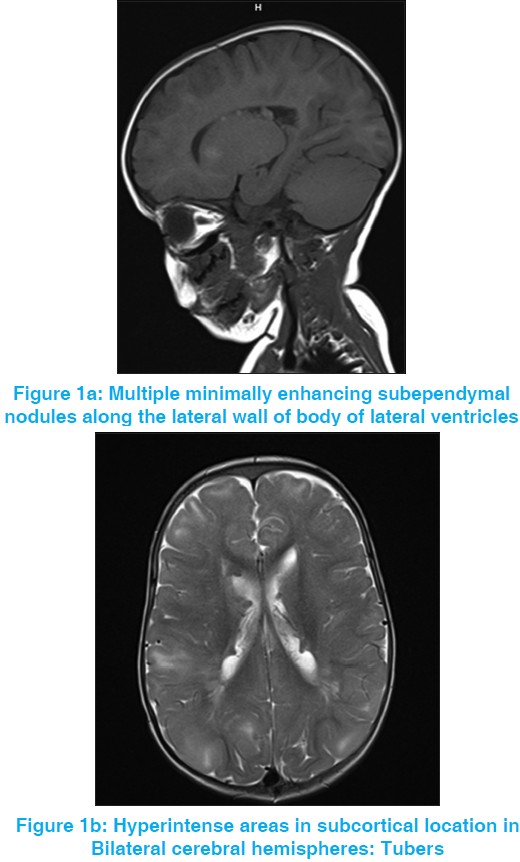 |
Tuberous sclerosis with rhabdomyoma |
p. 93 |
V Ajay, Vikram Singhal, Vardhelli Venkateshwarlu, SM Rajesh
DOI:10.4103/0971-6866.112912 PMID:23901201Tuberous sclerosis is a neurocutaneous syndrome characterized by abnormalities of both the integument and central nervous system. We present a case of tuberous sclerosis with rhabdomyoma in the heart. This was a 1½-year-old female child with infantile spasms and rhabdomyoma in heart with mother having neurocutaneous markers of tuberous sclerosis. Magnetic resonance imaging brain and electroencephalography findings were consistent with diagnosis. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
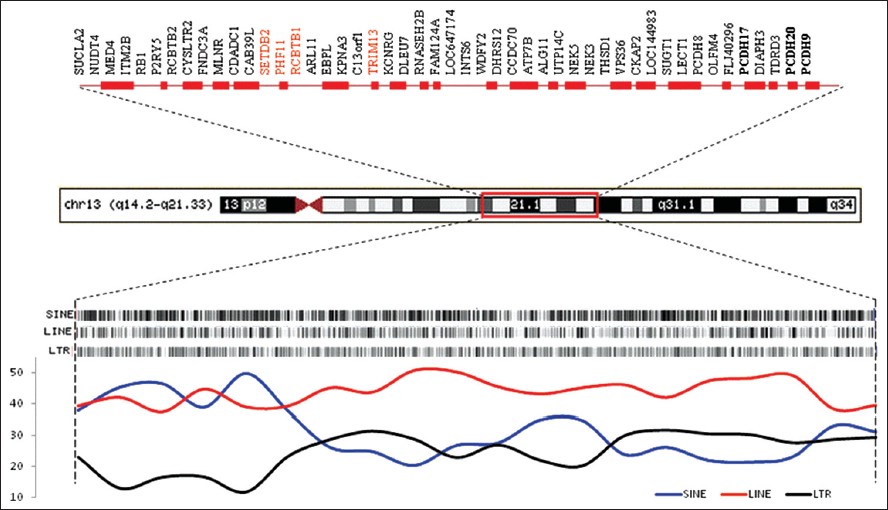 |
A novel deletion cluster at 13q14.2-q21.33 in an 80-year man with late onset leukemia: Clinical and molecular findings |
p. 96 |
Kiran Kumar, Sujit Maiti, Christina A Castellani, Richard O'Reilly, Shiva M Singh
DOI:10.4103/0971-6866.112916 PMID:23901202Chromosomal deletions are among the most common genetic events observed in hematologic malignancies; loss of genetic material is regarded as a hallmark of putative tumor suppressor gene localization. We have identified an unusual cluster of deletions at 13q14.2-13q21.33 in an 80-year-old father of a monozygotic twin pair discordant for schizophrenia, who developed chronic leukemia (CLL) at age 69.
Materials and Methods: The breakpoints for individual deletions in this cluster was identified by Affymetrix Human Array 6.0 screening.
Results: The deleted segments harbours a number of genes, most associated with cancer as well as a high concentration of LINEs, SINEs and related repeats. The derived chromosome represents an intra-chromosomal re-arrangement that quickly overtook blood progenitor cells probably before age 69 as a cause of CLL.
Conclusions: The study highlights the role of ongoing de novo changes at susceptible sites, such as repeat rich regions, in the human genome. Also, it argues for the involvement of genes/deletions in the 13q(14.2-21.33) region in the development of CCL. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
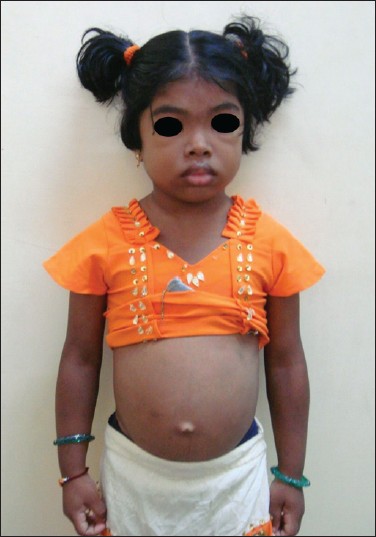 |
Short stature and an interesting association  |
p. 101 |
Latha Magatha Sneha, Kishore Thanasegarapandian, Venkataraman Paramasivam, Julius Xavier Scott
DOI:10.4103/0971-6866.112919 PMID:23901203Untreated hypothyroidism in children usually results in delayed puberty, but juvenile hypothyroidism causes isosexual precocious puberty in a rare syndrome called Van Wyk Grumbach syndrome, with a complete reversal to the pre pubertal state following thyroid hormone replacement therapy. We report here, a 7-year-old girl who presented with short stature, constipation and isosexual precocious puberty due to the long standing untreated severe hypothyroidism with this syndrome. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
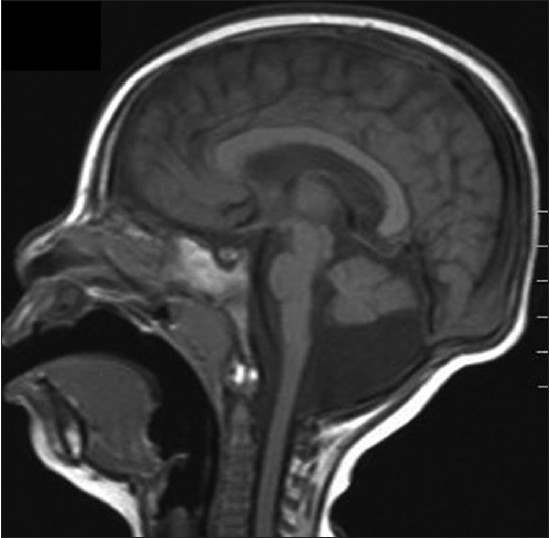 |
MICrocephaly, disproportionate pontine and cerebellar hypoplasia syndrome: A clinico-radiologic phenotype linked to calcium/calmodulin-dependent serine protein kinase gene mutation |
p. 104 |
Rashid Saleem, Gururaj Setty, Nahin Hussain
DOI:10.4103/0971-6866.112921 PMID:23901204MICrocephaly, disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome, a rare X-linked disorder, generally seen in girls, is characterized by neurodevelopmental delay, microcephaly, and disproportionate pontine and cerebellar hypoplasia. It is caused by inactivating calcium/calmodulin-dependent serine protein kinase (CASK) gene mutations. We report a 2-year-old girl with severe neurodevelopmental delay, microcephaly, minimal pontine hypoplasia, cerebellar hypoplasia, and normal looking corpus callosum, with whom the conventional cytogenetic studies turned out to be normal, and an array-comparative genomic hybridization (a-CGH) analysis showed CASK gene duplication at Xp11.4. Our case highlights the importance of using clinico-radiologic phenotype to guide genetic investigation and it also confirms the role of a-CGH analysis in establishing the genetic diagnosis of MICPCH syndrome, when conventional cytogenetic studies are inconclusive. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
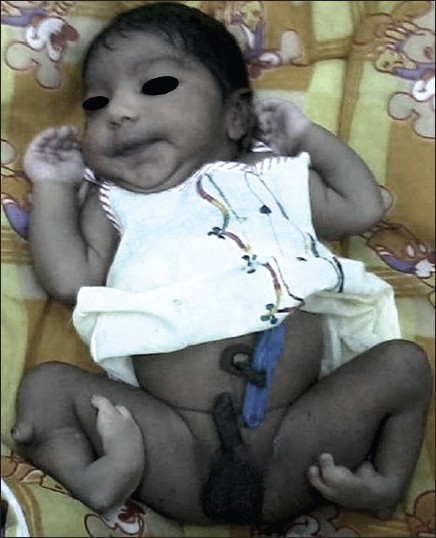 |
A case of bilateral tibial hemimelia type VIIa |
p. 108 |
Selvakumar Chinnakkannan, Rashmi Ranjan Das, K Rughmini, Sufath Ahmed
DOI:10.4103/0971-6866.112924 PMID:23901205Congenital absence of tibia is a rare anomaly, and may be total or partial, unilateral or bilateral. Total absence is more frequent than partial, unilateral absence occurs more often than bilateral, with right limb more commonly affected than the left. In partial defect, almost always the distal end of the bone is affected, and of the bilateral cases, there may be total absence on both sides, or total on one side and partial on the other. Males are slightly more commonly affected than the females. Though, the family history is usually negative for congenital abnormalities and other diseases, there is a considerable chance of occurrence of congenital defect of the tibia or of other abnormalities, in near or remote relatives. We report a case of newborn having bilateral tibial hemimelia type VIIa. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Triple X Egyptian woman and a Down's syndrome offspring |
p. 111 |
Faeza Abdel Mogib El-Dahtory
DOI:10.4103/0971-6866.112925 PMID:23901206The 47, XXX karyotype (triple X) has a frequency of 1 in 1000 female newborns. However, this karyotype is not usually suspected at birth or childhood. Female patients with a sex chromosome abnormality may be fertile. In patients with a 47, XXX cell line there appears to be an increased risk of a cytogenetically abnormal child but the extent of this risk cannot yet be determined; it is probably lower in the non-mosaic 47, XXX patient than the mosaic 46, XX/47, XXX one. We describe a new rare case of triple X woman and a Down's syndrome offspring. The patient is 26 years of age. She is a housewife, her height is 160 cm and weight is 68 kg and her physical features and mentality are normal. She has had one pregnancy at the age of 25 years resulted in a girl with Down's syndrome. The child had 47 chromosomes with trisomy 21 (47, XX, +21) [Figure 1]. The patient also has 47 chromosomes with a triple X karyotype (47, XX, +X) [Figure 2]. The patient's husband (27 years old) is physically and mentally normal. He has 46 chromosomes with a normal XY karyotype (46, XY). There are neither Consanguinity between her parent's nor she and her husband. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
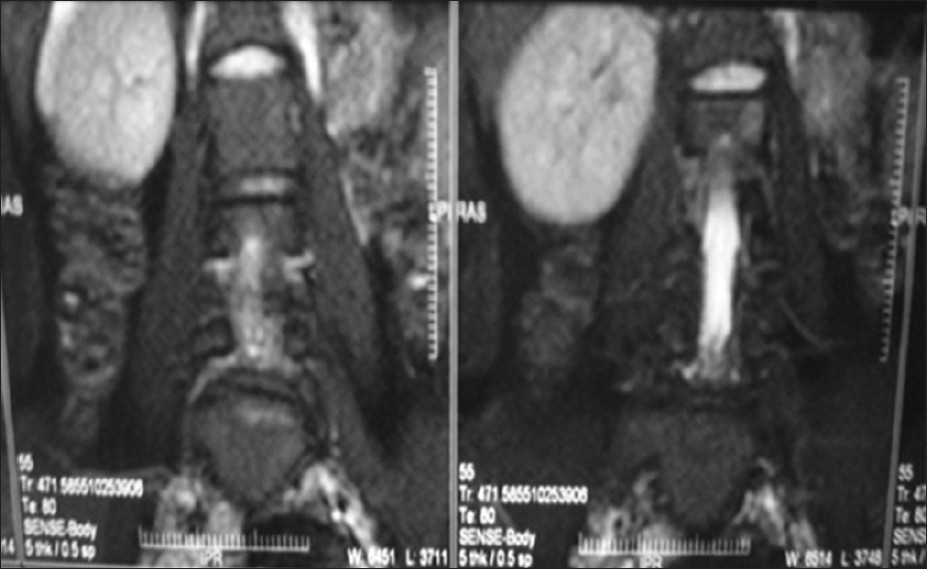 |
Mayer-Rokitansky-Kuster-Hauser syndrome type II: A rare case |
p. 113 |
Anand Pai, Mohammad Shakir
DOI:10.4103/0971-6866.112928 PMID:23901207Mayer-Rokitansky-Kuster-Hauser (MRKH) is a malformation complex comprising absent vagina and absent or rudimentary uterus. MRKH syndrome may be attributed to an initial affection of the intermediate mesoderm consequently leading (by the end of the 4 th week of fetal life) to an alteration of the blastema of the cervicothoracicsomites and the pronephricducts. These latter subsequently induce the differentiation of the mesonephric and then the Wolffian and Mullerian ducts. There are very sparse such cases reported. We present a case of type II MRKH or Mullerian renal cervical somite association (i.e., Mullerian duct aplasia, renal dysplasia, and cervical somite anomalies). |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| LETTER TO THE EDITOR |
|
|
|
|
Duchenne muscular dystrophy: Advances in molecular appraoch |
p. 116 |
Afaf Benitto, Khalil Hamzi, Mohammed Itri, Ilham Slassi, Sellama Nadifi
DOI:10.4103/0971-6866.112931 PMID:23901208 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|