|
Show all abstracts Show selected abstracts Add to my list |
|
| EDITORIAL |
|
|
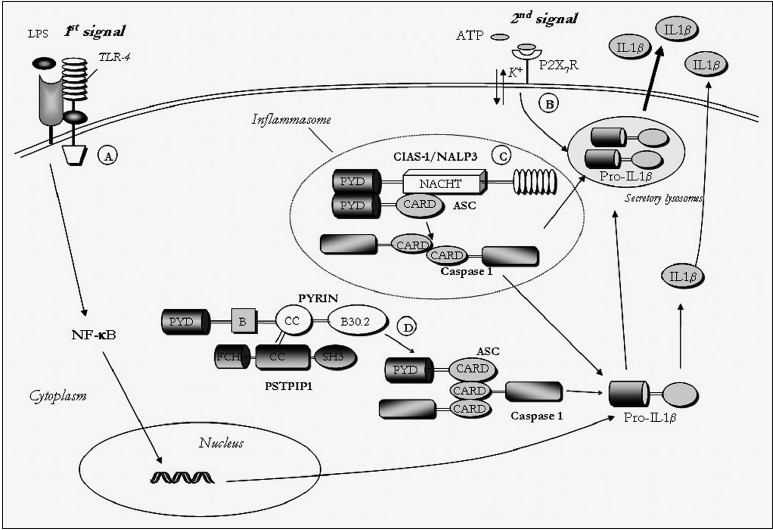  |
Familial Mediterranean fever: An unusual disease enlightening the inflammation biology |
p. 1 |
Kanjaksha Ghosh, Ajit C Gorakshakar
DOI:10.4103/0971-6866.132741 PMID:24959007 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| REVIEW ARTICLE |
 |
|
|
|
Genes and oral cancer |
p. 4 |
Sunit Kumar Jurel, Durga Shanker Gupta, Raghuwar D. Singh, Mrinalini Singh, Shilpi Srivastava
DOI:10.4103/0971-6866.132745 PMID:24959008Oral cancers have been one of the leading causes of deaths particularly in the developing countries. Prime reason for this high mortality and morbidity is attributed to the delay in diagnosis and prompt treatment. Relentless research in the field of oncology has led to the advent of novel procedures for the early detection of oral cancers. Molecular biology is highly promising in this regard. It is a procedure that detects alterations at a molecular level much before they are seen under a microscope and much before clinical changes occur. Molecular studies serve as the basis by which we will eventually be able not only to augment clinical assessment and classification of oral lesions but also predict malignant potential of oral lesions, thus reducing the incidence and increasing the scope for early diagnosis and treatment of oral cancers. However, making such sophisticated tools available for the common man in developing countries is one of the most important challenges faced today. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
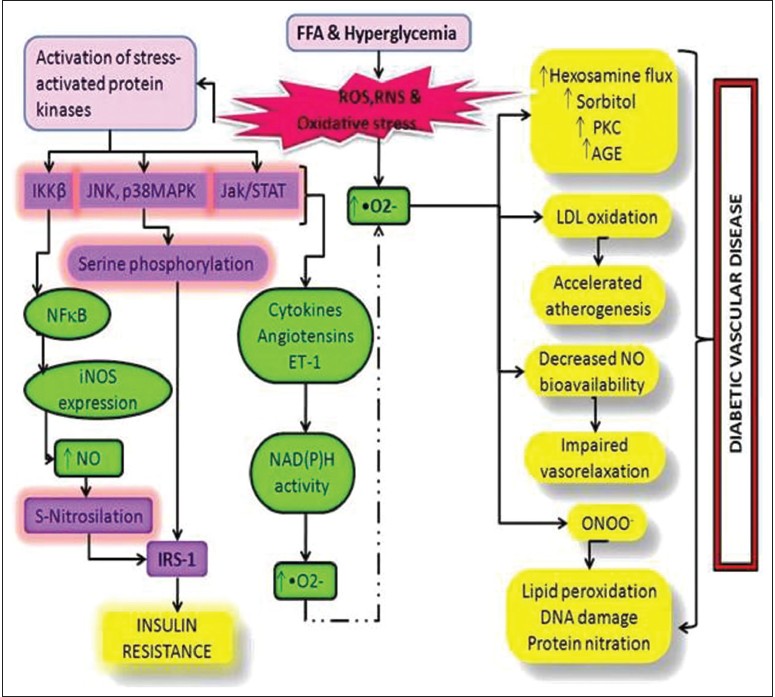 |
Reactive metabolites and antioxidant gene polymorphisms in type 2 diabetes mellitus  |
p. 10 |
Monisha Banerjee, Pushpank Vats
DOI:10.4103/0971-6866.132747 PMID:24959009Type 2 diabetes mellitus (T2DM), by definition is a heterogeneous, multifactorial, polygenic syndrome which results from insulin receptor (IR) dysfunction. It is an outcome of oxidative stress caused by interactions of reactive metabolites (RMs) with lipids, proteins and other molecules of the human body. Production of RMs mainly superoxides (•O2− ) has been found in a variety of predominating cellular enzyme systems including nicotinamide adenine dinucleotide phosphate oxidase, xanthine oxidase, cyclooxygenase, endothelial nitric oxide synthase (eNOS) and myeloperoxidase. The four main RM related molecular mechanisms are: increased polyol pathway flux; increased advanced glycation end-product formation; activation of protein kinase C isoforms and increased hexosamine pathway flux which have been implicated in glucose-mediated vascular damage. Superoxide dismutase, catalase, glutathione peroxidase, glutathione-S-transferase and NOS are antioxidant enzymes involved in scavenging RMs in normal individuals. Functional polymorphisms of these antioxidant enzymes have been reported to be involved in the pathogenesis of T2DM. The low levels of antioxidant enzymes or their non-functionality results in excessive RMs which initiates stress related pathways thereby leading to IR and T2DM. An attempt has been made to review the role of RMs and antioxidant enzymes in oxidative stress resulting in T2DM. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (3) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
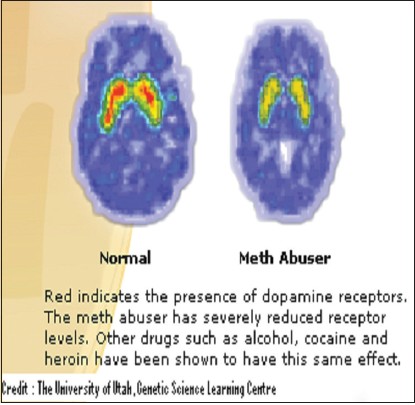 |
Neurotransmitters in alcoholism: A review of neurobiological and genetic studies |
p. 20 |
Niladri Banerjee
DOI:10.4103/0971-6866.132750 PMID:24959010Recent advances in the study of alcoholism have thrown light on the involvement of various neurotransmitters in the phenomenon of alcohol addiction. Various neurotransmitters have been implicated in alcohol addiction due to their imbalance in the brain, which could be either due to their excess activity or inhibition. This review paper aims to consolidate and to summarize some of the recent papers which have been published in this regard. The review paper will give an overview of the neurobiology of alcohol addiction, followed by detailed reviews of some of the recent papers published in the context of the genetics of alcohol addiction. Furthermore, the author hopes that the present text will be found useful to novices and experts alike in the field of neurotransmitters in alcoholism. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| ORIGINAL ARTICLES |
|
|
|
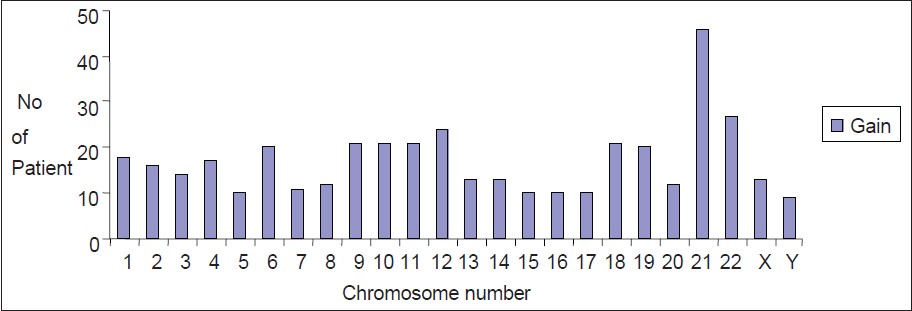 |
Pattern of chromosome involvement in childhood hyperdiploid pre-B-cell acute lymhoblastic leukemia cases from India |
p. 32 |
Lily S. Kerketta, Vundinti Baburao, Kanjaksha Ghosh
DOI:10.4103/0971-6866.132751 PMID:24959011Background: Hyperdiploid pre-B-cell acute lymhoblastic leukemia (pre-B-ALL) is a common form of childhood leukemia with very good prognosis with present day chemotherapy. However, the chromosomal composition of the hyperdiploidy has not been extensively studied and possible mechanism for this pathology remains so far conjectural.
Objective: To analyze the pattern of chromosome involvement in a cohort of childhood hyperdiploid pre-B-ALL from India and from this pattern to develop an understanding on the causation of such pathology. Whether such patients also carry translocations and FLT3 mutations in addition to their hyperdiploid karyotype.
Materials and Methods: One hundred and twenty-six childhood pre-B-ALL patients were studied. Bone marrow aspirate of these patients were evacuated for morphology, FAB classification, immunophenotyping and both conventional and molecular cytogenetics.
Results: Of 126 patients with pre-B-ALL (age 2-15 years), 90 patients with abnormal karyotype showed 50 with hyperdiploid karyotype (50/90 i.e. 55.5%). Chromosomes 9, 10, 14, 17, 18, 20 and 21 were more often involved in hyperdiploidy. Chromosome 21 duplication was present in 92% of the cases. Chromosomes 5, 15, 16, 17 and Y were less often involved (18-20%) in hyperdiploidy. About 44% of patients with hyperdiploidy had additional karyotypic abnormality of which TEL-AML1 was predominant (24%). Chromosome loss was rare and accounted for 20% of the cases only. We did not find any FLT3 mutation in our patients.
Conclusion: In this study, the pattern of chromosome involvement in hyperdiploid karyotype of ALL patients is same as other studies except some chromosomes like 1, 6, 11, 12, 19 and 22 have some more frequent involvement than other studies. This study also showed the occurrence of TEL/AML1 fusion is more (19.8%) than other reports from India. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
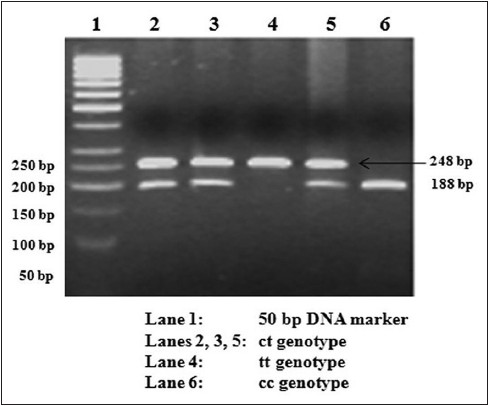 |
Association study of the ABCC8 gene variants with type 2 diabetes in south Indians |
p. 37 |
Radha Venkatesan, Dhanasekaran Bodhini, Nagarajan Narayani, Viswanathan Mohan
DOI:10.4103/0971-6866.132752 PMID:24959012Background: The ABCC8 gene which encodes the sulfonylurea receptor plays a major role in insulin secretion and is a potential candidate for type 2 diabetes. The -3c → t (rs1799854) and Thr759Thr (C → T, rs1801261) single nucleotide polymorphisms (SNPs) of the ABCC8 gene have been associated with type 2 diabetes in many populations. The present study was designed to investigate the association of these two SNPs in an Asian Indian population from south India.
Materials and Methods: A total of 1,300 subjects, 663 normal glucose tolerant (NGT) and 637 type 2 diabetic subjects were randomly selected from the Chennai Urban Rural Epidemiology Study (CURES). The -3c → t and Thr759Thr were genotyped in these subjects using polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) and a few variants were confirmed by direct sequencing.
Results: The frequency of the 't' allele of the -3c → t SNP was found to be 0.27 in NGT and 0.29 in type 2 diabetic subjects (P = 0.44). There was no significant difference in the genotypic frequency between the NGT and type 2 diabetic group (P = 0.18). Neither the genotypic frequency nor the allele frequency of the Thr759Thr polymorphism was found to differ significantly between the NGT and type 2 diabetic groups.
Conclusion: The -3c → t and the Thr759Thr polymorphisms of the ABCC8 gene were not associated with type 2 diabetes in this study. However, an effect of these genetic variants on specific unidentified sub groups of type 2 diabetes cannot be excluded. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
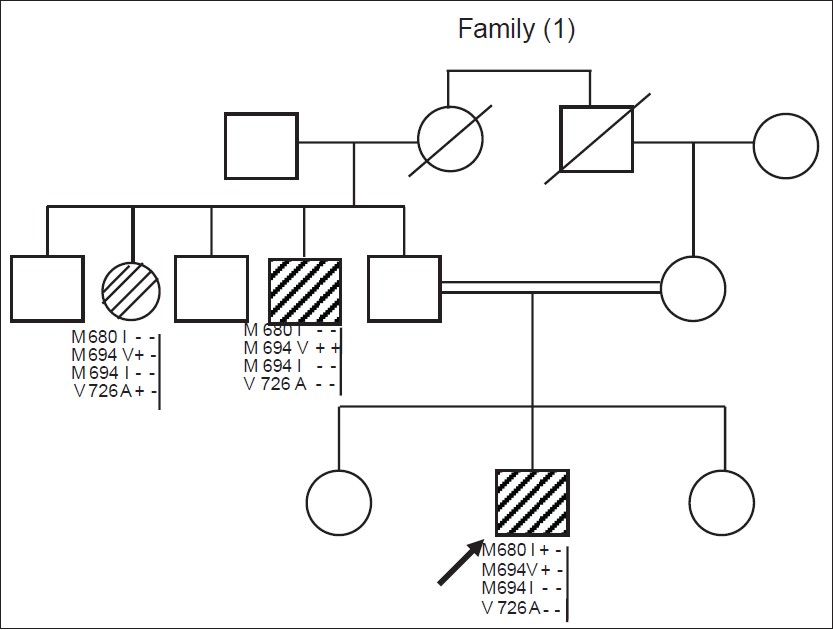 |
Phenotype-genotype updates from familial Mediterranean fever database registry of Mansoura University Children' Hospital, Mansoura, Egypt |
p. 43 |
Mohammad S. Al-Haggar, Sohier Yahia, Dina Abdel-Hady, Afaf Al-Saied, Rasha Al-Kenawy, Rabab Abo-El-Kasem
DOI:10.4103/0971-6866.132755 PMID:24959013Background: Familial Mediterranean fever (FMF) is autosomal recessive disease that affects people from Mediterranean region, Europe and Japan. Its gene (Mediterranean fever [MEFV]) has more than 100 mostly non-sense mutations.
Objectives: The objective of the following study is to provide some phenotype-genotype correlates in FMF by categorizing the Egyptian FMF cases from Delta governorates after analysis of the four most common mutations of MEFV gene (M680I, M694I, M694V, V726A).
Subjects And Methods: Clinically, suspected FMF cases using Tel-Hashomer criteria were enrolled in the study. Cases were referred to Mansoura University Children's Hospital that serves most of the most middle Delta governorates, in the period from 2006 to 2011. Subjects included 282 males and 144 females, mean age of onset 9.3 ± 2.2 years. All cases were analyzed for these mutations using amplification refractory mutation system based on the polymerase chain reaction technique. Five FMF patients agreed to undergo renal biopsy to check for development of amyloidosis. Analysis of data was carried out using SPSS (SPSS, Inc., Chicago, IL, USA).
Results: Mutation was found in 521 out of 852 studies alleles, the most frequent is M694V (35.4%) followed by M694I, V726A and M680I. 11 cases were homozygous; 7 M694V, 3 M680I and only one M694I case. Severe abdominal pain occurred in 31 (7.28%) but severe arthritis in 103 cases (24.2%). Strong association was found between arthritis and homozygous mutant compared with single and double heterozygous (72.7% vs. 33.3% and 20.24%, P < 0.001). Four amyloid cases were M694V positive. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
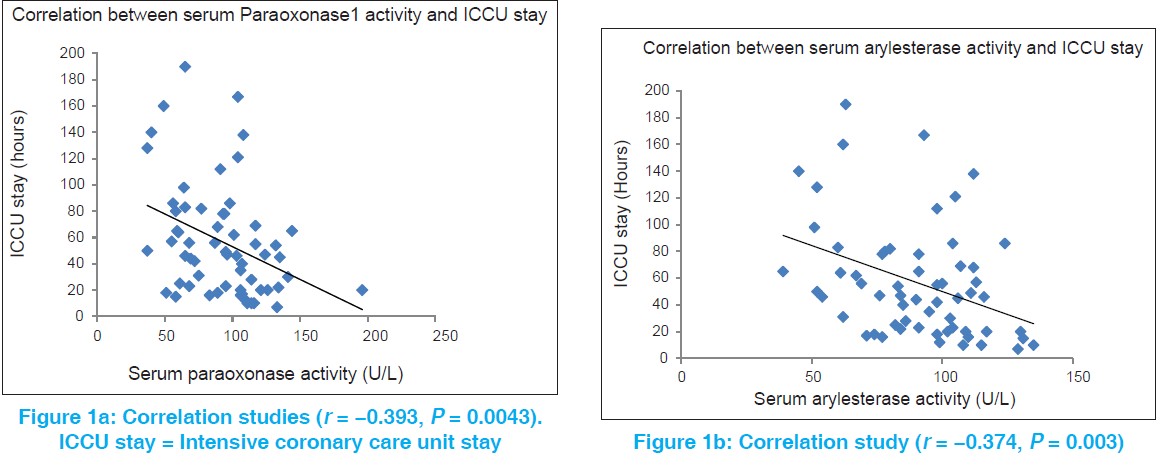 |
Paraoxonase1, its Q192R polymorphism and HDL-cholesterol in relation to intensive cardiac care unit stay in ischemic heart disease |
p. 51 |
Mahesh Harishchandra Hampe, Mukund Ramchandra Mogarekar
DOI:10.4103/0971-6866.132756 PMID:24959014Aims And Objectives: The present study was evaluated the atheroprotective potential of paraoxonase1 (PON1) and its Q192R polymorphism, to determine whether this polymorphism, which is responsible for differential PON1 activity plays any role in the pathogenesis, severity and extent of coronary artery disease (CAD).
Materials and Methods: This hospital-based cross-sectional study investigated 60 diagnosed cases of CAD and 60 age and gender matched controls. All were assessed for serum PON1 activity, PON1 Q192R polymorphism and for classical cardiovascular risk factors. Individual serum phenotyping for PON1 Q192R polymorphism was determined by double substrate hydrolysis assay. Severity of CAD was assessed by the length of intensive cardiac care unit (ICCU) stay.
Results: Serum PON1 activity is significantly reduced in cases of CAD (92.6 ± 31.13 IU/L when compared with controls (105.26 ± 32.53 IU/L). Furthermore, serum arylesterase activity is reduced in CAD patients (90.31 ± 23.26 kU) when compared with the control subjects (101.61 ± 28.68 kU). Serum PON1 and arylesterase activities are significantly negatively correlated with the length of ICCU stay (r = −393 and r = −374 respectively). There is no significant difference in the occurrence of CAD and length of ICCU stay among the PON1 phenotypes (P = 0.92). Logistic regression analysis after adjustment of established risk factors revealed no significant association between CAD risk and PON1 Q192R polymorphism (odds ratios: 1.179 [95% confidence intervals: 0.507-2.744], P = 0.702).
Summary And Conclusions: The current study demonstrates that the activity of the PON1 enzyme may be more important factor than the PON1 Q192R polymorphism in the severity and extent of CAD. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Association of single nucleotide polymorphisms of CACNA1A gene in migraine |
p. 59 |
Aadil Bashir, Shiekh Saleem, Maqbool Wani, Roohi Rasool, Irfan Yousuf Wani, Azhara Gulnar, Sawan Verma
DOI:10.4103/0971-6866.132757 PMID:24959015Introduction: Migraine is a chronic, neurovascular polygenic disease where genetic and environmental factors are involved in its etiology. Dysfunction of neuronal ion transportation can provide a model for predisposition for common forms of migraine. Mutations in genes encoding ion channels disturb the rhythmic function of exposed tissue that may also explain the episodic nature of migraine. Our aim was to study the single nucleotide polymorphisms of CACNA1A gene in migraine patients.
Materials and Methods: The subjects were the patients of migraine, in the age range of 18-80 years, diagnosed by a Neurologist, as per the diagnostic criteria of International Headache Society (IHS) Classification 2004 after excluding other causes of headache by clinical examination and relevant investigations.
The controls were the age and sex matched healthy persons from the same population excluding the relatives of patients. Only those patients and the controls, who voluntarily participated in the study, were taken and their blood samples were taken for the study. Deoxyribonucleic acid (DNA) extraction was performed according to the manufacturer's protocol for Qiagen DNA extraction kits (Qiagen, Hilden, NRW, Germany). DNA content was quantified by spectrophotometric absorption (Nanodrop Spectrophotometer, BioLab, Scoresby, VIC, Australia). Polymerase chain reaction was performed using an iCycler Thermal Cycler (Bio-Rad, Hercules, CA, USA). The polymorphic analysis of CACNA1A gene was carried out by two methods: Restriction fragment length polymorphism and sequencing.
Results: The study included a total of 25 patients of migraine, diagnosed on out-patient department basis as per IHS Classification 2004 and compared with age and sex matched 25 healthy controls. Most of the patients 23 (92%) were below the age of 50 years. 20 of the patients (80%) were females and 5 (20%) were males. The polymorphic analysis of CACNA1A gene revealed the presence of only the wild form of the gene for the codon E993V in both case and control groups.
Conclusion: In our study, we could not find any polymorphism of CACNA1A gene in the selected patients. Instead the wild type of genotype was found in both patients and controls. This negative result presented here, implies that if the CACNA1A gene is involved in typical migraine (with and without aura), its contribution is very modest and therefore difficult to discern. Nevertheless, there are other genes that could be considered potential candidates for typical migraine susceptibility for which further research is needed. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
A comprehensive analysis of breakpoint cluster region-abelson fusion oncogene splice variants in chronic myeloid leukemia and their correlation with disease biology |
p. 64 |
Zafar Iqbal
DOI:10.4103/0971-6866.132758 PMID:24959016Background: BCR-ABL fusion oncogene is a hallmark of Chronic Myeloid Leukemia (CML). It results due to translocation between chromosome 22 and chromosome 9 [t (9; 22)(q34; q11)]. It gives rise to translation of a 210 KDa chimeric protein (p210), leading to enhanced tyrosine kinase activity and activation of leukemogenic pathways, ultimately causing onset of CML. In case of CML, the classic fusions are b2a2 or b3a2, fusing exon 13 (b2) or exon 14 (b3) of BCR, respectively, to exon 2 (a2) of ABL. The type of BCR-ABL transcripts are thought to be have different prognosis and hence useful in clinical decision-making. The frequencies of different fusion oncogenes associated with leukemia can vary in different ethnic groups and geographical regions due to interplay of genetic variation in different ethnic populations, diverse environmental factors and living style. Moreover, earlier relevant studies from our region were carried out in small subset of patients. Therefore, objective of this study was to find out frequencies of different BCR-ABL splice variants in larger subset of CML patients.
Methods: A nested reverse transcriptase polymerase chain reaction (RT-PCR) was established to detect BCR-ABL splice variants in 130 CML patients. Sensitivity of RT-PCR and ability to detect BCR-ABL fusion gene in least possible time was studied.
Results: BCR-ABL detection using our optimized RT-PCR protocol could be completed in 8 hours, starting from RNA extraction to Gel electrophoresis. Sensitivity of RT-PCR assay was of the order of 10−6 . Out of 130 Pakistani patients, 83 (63.84%) expressed b3a2 while 47 (36.15%) expressed b2a2 transcript.
Conclusion: Our RT-PCR was proved to be very quick to detect BCR-ABL fusion oncogene in CML patients within one working day. Because of its sensitivity, it can be used to monitor complete molecular response in CML. BCR-ABL RT-PCR and BCR-ABL splice variants frequency in our study differs from other ethnic groups. It shows that ethnic and geographical differences exist in BCR-ABL splice variant frequency, which may have a profound effect on disease biology as well as implications in prognosis and clinical management of BCR-ABL positive leukemias. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| CASE REPORTS |
|
|
|
 |
Chronic myeloid leukemia in case of Klinefelter syndrome |
p. 69 |
Vasundhara Chennuri, Rajesh Kashyap, Parag Tamhankar, Subha Phadke
DOI:10.4103/0971-6866.132760 PMID:24959017Klinefelter syndrome (KS) is a sex chromosome disorder and has been reported to be associated with increased risk for malignancies. We report a 22-year-old male patient who was diagnosed to have chronic myeloid leukemia in chronic phase. Bone marrow cytogenetic examination revealed karyotype 47, XXY, t (9; 22)(q34, q11) suggestive of KS with presence of Philadelphia chromosome. The patient was treated with oral imatinib mesylate (400 mg/day). Complete hematological response was achieved after 2 months of therapy. The bcr-abl/abl transcript percentage measured from peripheral blood at baseline, 1 and 2 years after imatinib were 97%, 1.99%, 0.007%, respectively. He remains in complete hematological and major molecular remission after 2 years of continued imatinib therapy. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
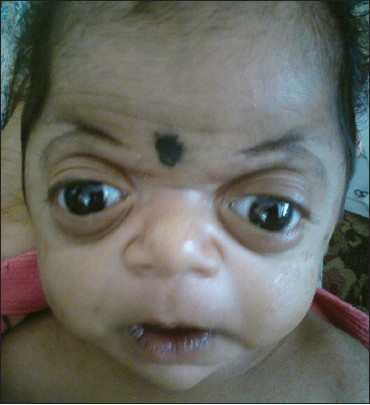 |
Raine syndrome |
p. 72 |
B. Vishwanath, K. Srinivasa, M. Veera Shankar
DOI:10.4103/0971-6866.132761 PMID:24959018Raine syndrome is a rare genetic disorder with characteristic features of exophthalmos, choanal atresia or stenosis, osteosclerosis and cerebral calcifications. Most of babies with this disorder die immediately after birth. We report a baby who was 7 weeks old at the time of presentation. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Berardinelli-Seip syndrome type 1 in an Egyptian child |
p. 75 |
Kotb Abbass Metwalley, Hekma Saad Farghaly
DOI:10.4103/0971-6866.132762 PMID:24959019Berardinelli-Seip syndrome type 1 or Berardinelli-Seip congenital lipodystrophy 1 (BSCL1) is a very rare genetic disorder characterized by lipoatrophy, hypertriglyceridemia, hepatomegaly and acromegaloid features. Its prevalence in Egypt is not known. Here, we report case of a 12-year-old Egyptian boy with the clinical, metabolic and molecular genetics manifestations of BSCL1 including overt diabetes mellitus. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
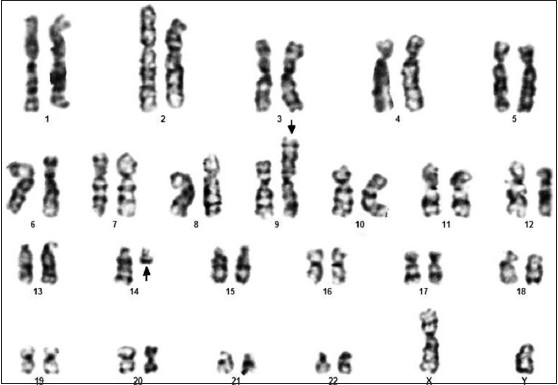 |
A novel chromosomal abnormality t (9;14)(p24;q13) in B-acute lymphoblastic leukemia |
p. 79 |
Sureshkumar Raveendran, Santhi Sarojam, Geetha Narayanan, Hariharan Sreedharan
DOI:10.4103/0971-6866.132763 PMID:24959020Acute lymphoblastic leukemia is a malignant disease of the bone marrow in which early lymphoid precursors proliferate and replace the normal hematopoietic cells of the marrow. We describe the clinical, morphologic, immunophenotypic and cytogenetic findings in the case of a 26-year-old man with B-lymphoblastic leukemia. Surface marker analysis revealed that they are positive for CD markers CD10, CD19, CD13, CD34, CD45 and HLA-DR, but negative for CD20, CD33, CD117 and CD11C markers. Cytogenetic analysis established a novel translocation, t (9;14)(p24;q13). Apart from this, spectral karyotyping revealed an additional translocation, t (6p; 14q). This is the first documented case of B-lymphoblastic leukemia with concurrent occurrence of both abnormalities. Further studies are needed to understand the role of this abnormality in carcinogenesis. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
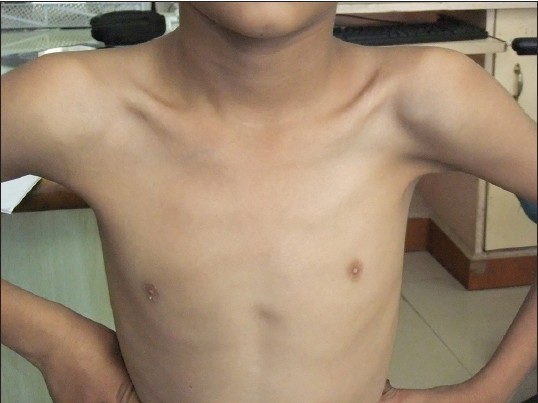 |
Poland syndrome |
p. 82 |
Chandra Madhur Sharma, Shrawan Kumar, Manoj K. Meghwani, Ravi P. Agrawal
DOI:10.4103/0971-6866.132764 PMID:24959021Poland's syndrome is a rare congenital condition, characterized by the absence of the sternal or breastbone portion of the pectoralis major muscle, which may be associated with the absence of nearby musculoskeletal structures. We hereby report an 8-year-old boy with typical features of Poland syndrome, the first documented case from Uttar Pradesh, India. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
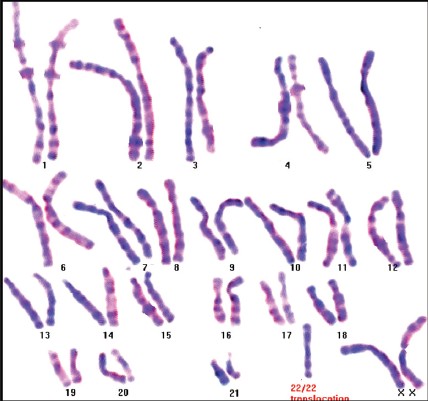 |
Unexpected Inheritance of a Balanced Homologous translocation t(22q;22q) from father to a phenotypically normal daughter |
p. 85 |
D. K. Chopade, Harish Harde, Pallavi Ugale, Sandesh Chopade
DOI:10.4103/0971-6866.132765 PMID:24959022Rearrangements between homologous chromosomes are extremely rare and manifest mainly as monosomic or trisomic offsprings. There are remarkably few reports of balanced homologous chromosomal translocation t (22q; 22q) and only two cases of transmission of this balanced homohologous rearrangement from mother to normal daughter are reported. Robersonian translocation carriers in non-homologous chromosomes have the ability to have an unaffected child. However, it is not possible to have an unaffected child in cases with Robersonian translocations in homologous chromosomes. Carriers of homologous chromosome 22 translocations with maternal uniparental disomy do not have any impact on their phenotype. We are presenting a family with a history of multiple first trimester miscarriages and an unexpected inheritance of balanced homologous translocation of chromosome 22 with paternal uniparental disomy. There are no data available regarding the impact of paternal UPD 22 on the phenotype. We claim this to be the first report explaining that paternal UPD 22 does not impact the phenotype. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
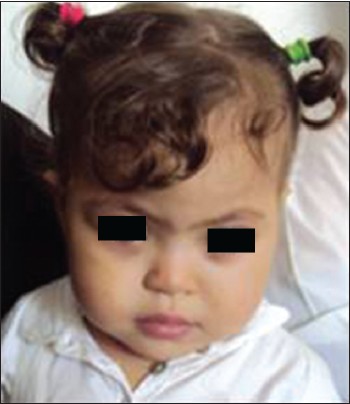 |
Characterization of a rare short arm heteromorphism of chromosome 22 in a girl with down-syndrome like facies |
p. 89 |
Abdelhafid Natiq, Siham Chafai Elalaoui, Thomas Liehr, Saïd Amzazi, Abdelaziz Sefiani
DOI:10.4103/0971-6866.132767 PMID:24959023Chromosomal heteromorphisms are described as interindividual variation of chromosomes without phenotypic consequence. Chromosomal polymorphisms detected include most regions of heterochromatin of chromosomes 1, 9, 16 and Y and the short arms of all acrocentric chromosomes. Here, we report a girl with Down-syndrome such as facies and tremendously enlarged short arm of a chromosome 22. Fluorescence in situ hybridization (FISH) with a probe specific for all acrocentric short arms revealed that the enlargement p arms of the chromosome 22 in question contained exclusively heterochromatic material derived from an acrocentric short arm. Parental studies identified a maternal origin of this heteromorphism. Cryptic trisomy 21 of the Down-syndrome critical region was excluded by a corresponding FISH-probe. Here, we report, to the best of our knowledge, largest ever seen chromosome 22 short arm, being ~×1.5 larger than the normal long arm. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| BRIEF REPORTS |
|
|
|
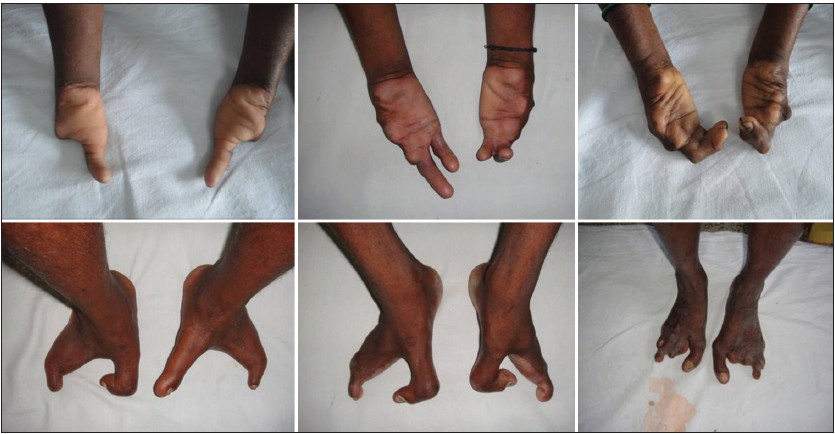 |
Split-hand/feet malformation in three tamilian families and review of the reports from India |
p. 92 |
S. Deepak Amalnath, Maya Gopalakrishnan, Tarun Kumar Dutta
DOI:10.4103/0971-6866.132769 PMID:24959024Split-hand/foot malformation (SHFM) is a rare condition which can be either syndromic or nonsyndromic. We report three unrelated pedigrees, one with autosomal dominant (AD) inheritance and the other two with autosomal recessive (AR) pattern. We also briefly review the published reports from India. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| LETTER TO EDITOR |
|
|
|
|
Glucose 6-phosphate dehydrogenase deficiency in Muslim community settled in Jaunpur district |
p. 96 |
Vandana Rai, Pradeep Kumar
DOI:10.4103/0971-6866.132770 PMID:24959025 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| LETTERS TO EDITOR |
|
|
|
|
ß-thalassemia and alkaptonuria |
p. 97 |
Sora Yasri, Viroj Wiwanitkit
DOI:10.4103/0971-6866.132772 PMID:24959026 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|