 |
|
Médicaments sur prescription, responsabilité des produits, et préemption de responsabilité civile
Catherine D. DeAngelis, MD, MPH;
Phil B. Fontanarosa, MD, MBA
JAMA. 2008;300(16):1939-1941
Les bénéfices et risques associés aux agents pharmacologiques et aux appareillages médicaux ont reçu une attention croissante de la part de la communauté médicale, du public, et des agences gouvernementales. De sérieuses inquiétudes sur les capacités et la surveillance de la Food and Drug Administration (FDA) pour assurer une détermination précise de la sécurité et de l’efficacité, des mécanismes appropriés et des décisions concernant l’approbation des médicaments et des appareillages, et la surveillance effective post-commercialisation ont incité à faire des recherches par l’Institut de Médecine1 et par une commission FDA blue ribbon.2 Une législation majeure3 a été ordonnée pour améliorer la capacité de l’agence à accomplir sa responsabilité régulatrice à assurer que les produits pharmaceutiques et les appareillages médicaux approuvés pour la commercialisation sont sûrs.4
Bien que l’évaluation de nouveaux médicaments et appareillages soit techniquement rigoureuse, l’approche actuelle de baser les décisions d’approbation des médicaments sur les essais cliniques d’efficacité qui incluent de relativement petits nombres de patients garantit virtuellement que les risques totaux et un profil de sécurité complet ne sera pas identifié au moment de l’approbation.5 Plutôt, le profil total de sécurité ne se manifeste que sur une population très étendue de patients qui sont sélectionnés moins soigneusement que les participants à des essais cliniques.
L’article par Giezen et al6 dans ce numéro de JAMA est un exemple important de ce phénomène bien connu. Dans une analyse de 174 produits biologiques (ex, anticorps, hormones, enzymes) approuvés de janvier 1995 à juin 2007, incluant 136 agents approuvés aux Etats-Unis, 105 approuvés dans l’Union européenne, et 67 approuvés dans les deux continents, les auteurs ont trouvé que 82 actions régulatrices de sécurité (incluant 19 avertissements « black box ») étaient publiés pour 41 des 174 produits biologiques différents (23,6%). La probabilité d’un agent biologique ayant une action régulatrice de sécurité était de 14% à 3 ans et 29% à 10 ans après approbation, avec les premiers agents approuvés dans une classe biologique spécialement enclins à une action régulatrice de sécurité.
Etant donné le processus courant imparfait pour l’approbation et le système de surveillance de post-commercialisation défectueux, le processus de régulation des médicaments et des dispositifs est au mieux une science inexacte et incomplète. Jusqu’à ce que ces manques dans le système soient remédiées, certains patients continueront inévitablement à éprouver un aspect négatif de l’usage de produits récemment commercialisés aussi bien que de l’usage d’autres médications approuvées. De même qu’avec d’autres produits grand public qui peuvent causer des dommages, les consommateurs (i.e. les patients) qui sont lésés par des dispositifs médicaux défectueux ou par des produits pharmaceutiques avec des avertissements inadéquats sur les effets potentiels pourraient avoir recours à une action légale de recours. Selon Gostin,7 le litige et la loi de responsabilité civile de l’état « apporte un système de justice civile conçue pour compenser les patients, dissuader une conduite dangereuse, et encourager l’innovation dans la conception du produit, l’emballage, l’étiquetage, et la publicité. »7
Ainsi, la responsabilité civile sert en réalité de façon de combler le fossé régulateur dans le processus d’approbation de pré-commercialisation de la FDA et de fournir un mécanisme pour la surveillance post-commercialisation.7,8 De plus, le litige a été une source riche d’informations sur la façon dont les compagnies manufacturant les médicaments et les dispositifs se comportent, comme avec la promotion off-label (non conforme à l’AMM), paternité de l’écrit par un auteur invité ou un « nègre », et rapportant des informations sur la sécurité.9,10 Sans l’information révélée par la sortie publique de documents sur des actions de responsabilité civile, beaucoup de ces comportement resteraient inconnus, certains jugements de fabricants de médicaments sur les problèmes de sécurité seraient dissimulés, à l’examen ou à la surveillance, et la FDA ne pourrait pas le savoir non plus.
Un prochain important cas capital de la Cour Suprême, Wyeth vs Levine, déterminera si les patients doivent être autorisés à porter plainte contre les fabricants de produits pharmaceutiques pour les dommages résultant de « lacune au niveau des avertissements » des risques potentiels, bien que l’étiquette du produit et l’information de la notice d’utilisation aient été approuvées par la FDA. Le cas concerne une femme de 62 ans ayant reçu une injection intra-musculaire d’hydrochloride de prométhazine (Phenergan : Wyeth Pharmaceuticals Inc) puis un bolus intraveineux du médicament pour le traitement de symptômes liés à une migraine. Cependant, la seconde injection de médicament a censément impliqué un contact artériel, résultant en une atteinte vasculaire et une progression vers la gangrène, et, finalement, l’amputation de sa main et son avant-bras.
La Cour Suprême du Vermont avait confirmé une décision d’une Cour d’Appel ayant constaté que Wyeth était négligent et n’avait pas réussi à apporter un avertissement adéquat concernant les dangers connus de l’injection intra-vasculaire de prométhazine, et la cour a accordé à la patiente 6,8 millions de dollars.11 Wyeth lutte pour que les poursuites soient interdites parce que l’information de la notice d’utilisation avait été approuvée par la FDA. Il est question pour la Cour de savoir si l’approbation de la FDA dans l’étiquetage du médicament délivré sur ordonnance « anticipe » les réclamations de responsabilité légales concernant les produits contre un fabricant pharmaceutique dont les produits sont découverts plus tard comme présentant des risques de sécurité et causant des dommages. Spécifiquement, la question est de savoir si un fabricant qui observe les exigences d’avertissement de la FDA peut être tenu pour responsable en responsabilité civile de l’état pour ne pas avoir apporter des avertissements adéquats.
La préemption, connue aussi sous le terme de préemption fédérale, est l’une des questions les plus controversées de la loi américaine sur la responsabilité du fabricant. La préemption est le concept que la loi fédérale a la priorité sur la loi d’état ou locale, de telle sorte que le gouvernement fédéral peut empêcher les états de tout règlementer sur le même sujet. La prédominance fédérale tient à ce que la loi fédérale défende ou « anticipe » la réglementation d’état contradictoire lorsqu’existe une loi fédérale règlementant le sujet particulier.
La source du pouvoir fédéral de préemption se trouve dans la Clause de Suprématie de la Constitution des Etats-Unis, qui stipule que les lois fédérales « seront la Loi suprême du Pays ». Conformément à cette clause, les cours sont obligées de suivre une instruction équivoque dans une législation fédérale qui anticipe la loi d’état. Essentiellement, la préemption signifie que l’arrangement régulateur fédéral est la loi de direction et est l’une des défenses les plus communément utilisée dans la défense de la responsabilité des produits.
Un autre cas examinant la préemption de 2008, Riegel v Medtronic, 12 impliquait une réclamation de responsabilité d’un appareil médical par un patient qui cherchait des réparations pour dommages qui résultaient d'un cathéter d'angioplastie lors d’une procédure coronaire percutanée. La Cour Suprême a soutenu que les réclamations du patient dépendaient de l’amendement sur les appareillages médicaux de 1976 (MDA) 13 du 1938 Federal Food, Drug, and Cosmetic Act (FDCA).14
Le MDA barre effectivement le litige qui conteste la sûreté ou l'efficacité d'un médical dispositif qui a reçu l'approbation premarket de la FDA et est lancé sur le marché sous la forme qui a reçu cet approval.7 Le MDA la « condition » acquiert expressément n'importe quel état dont est « différent, ou en plus » de n'importe quelle condition fédérale ou laquelle « rapporter à la sûreté ou à l'efficacité du dispositif » sous law.7 fédéral juste après l'acte de Riegel, des avocats défendant les fabricants d'appareil médical ont déposé des motions dans l'état et les cours fédérales réclamations pour écarter plaignants des' sous preemption.15
Dans le cas prochain de Wyeth v Levine, avec des arguments d'ouverture programmé pour le novembre 2008, la court suprême déterminera si le FDCA, qui régit le règlement des agents pharmaceutiques et n'a aucune préemption exprès la clause, a impliqué la préemption du litige d'acte délictuel d'état. Juste comme avec le FDCA, les amendements récemment décrétés de FDA Act3 également n'inclut pas une disposition pour la préemption.
Néanmoins, alors que les positions précédentes de FDA ont responsabilité soutenue d'acte délictuel comme mécanisme important pour la drogue la sûreté, assurant « une couche distincte de protection des consommateurs », la FDA courante placent est une cette préemption de faveurs. Cependant, selon Kessler et Vladek,16 le FDAAA apparaît pour miner cette position courante de pro-préemption près codifiant les conditions existantes « qui obligent des fabricants de drogue à fournir des informations à jour de sûreté aux médecins et les patients et autorisent des fabricants à faire ainsi sans fixer d'abord l'approbation de la FDA. La codification de cet engagement dégage l'argument principal de pro-préemption la FDA et les fabricants font-à savoir, cela seule la FDA décide le contenu des étiquettes de drogue. »16 en conséquence, en réponse aux risques nouvellement découverts, fabricants de drogue avoir l'autorité et la responsabilité de modifier l'étiquetage quand les risques se manifestent et peuvent faire tellement en dehors fixant l'approbation préalable des risques FDA.16 découverts avant l'approbation de drogue ou après marketing de produit qui sont connu des investigateurs ou des fabricants de drogue doit être révélé à la FDA, au public, aux médecins, et aux pharmaciens aussi rapidement que possible, et il ne devrait y avoir aucune récompense ou incitation pour le retard. N'est-elle pas la préemption d'acte délictuel d'état juste cela ?
Informations sur les auteurs
| |
Liens financiers : Aucun déclaré.
Autres contributions : Nous remercions Joseph P. Thornton, JD (JAMA), Bruce M. Psaty, MD, PhD (University of Washington, Seattle), and Lawrence O. Gostin, JD (Georgetown Law Center, Washington, DC), pour leurs commentaires utiles et précieux dans la revue de cet article. Aucun d’entre eux n’a reçu de compensation financière pour cette revue.
Les éditoriaux représentent les opinions des auteurs et du JAMA et pas celle de l’American Medical Association.
Affiliations des auteurs: Dr DeAngelis is Editor in Chief et Dr Fontanarosa (phil.fontanarosa{at}jama-archives.org) is Executive Deputy Editor, JAMA.
Voir aussi p 1887.
BIBLIOGRAPHIE
| |
1. Institute of Medicine. The Future of Drug Safety: Promoting and Protecting the Health of the Public. Washington, DC: National Academies Press; 2006.
2. Food and Drug Administration. Science Board. Science and Mission at Risk: Report of the Subcommittee on Science and Technology. Washington, DC: FDA; 2007.3. FDA Amendments Act (FDAAA) of 2007, Pub L No. 110-85, 121 Stat 823.4. Psaty BM, Korn D. Congress responds to the IOM Drug Safety Report—in full. JAMA. 2007;298(18):2185-2187.
FREE FULL TEXT
5. Fontanarosa PB, Rennie D, DeAngelis CD. Postmarketing surveillance—lack of vigilance, lack of trust. JAMA. 2004;292(21):2647-2650.
FREE FULL TEXT
6. Giezen TJ, Mantel-Teeuwisse AK, Straus SMJM, et al.. Safety-related regulatory actions for biologicals approved in the United States and the European Union. JAMA. 2008;300(16):1887-1896.
FREE FULL TEXT
7. Gostin LO. The deregulatory effects of preempting tort litigation: FDA regulation of medical devices. JAMA. 2008;299(19):2313-2316.
FREE FULL TEXT
8. Kesselheim AS, Avorn J. The role of litigation in defining drug risks. JAMA. 2007;297(3):308-311.
FREE FULL TEXT
9. Psaty BM, Kronmal RA. Reporting mortality findings in trials of rofecoxib for Alzheimer disease or cognitive impairment: a case study based on documents from rofecoxib litigation. JAMA. 2008;299(15):1813-1817.
FREE FULL TEXT
10. Ross JS, Hill KP, Egilmn DS, Krumholz HM. Guest authroship and ghostwriting in publications related to rofecoxib: a case study of industry documents from rofecoxib litigation. JAMA. 2008;299(15):1800-1812.
FREE FULL TEXT
11. Levine v Wyeth, 2006 VT 107 (Vt 2006), cert granted, 128 SCt 1118 (2008).12. Riegel v Medtronic Inc, 128 SCt 999 (2008).13. Medical Device Amendments (MDA), 21 USC 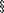 360k(a) (1976).14. Federal Food, Drug, and Cosmetic Act, 21 USC 301 et seq.15. Feder BJ. Medical device ruling redraws lines on lawsuits. New York Times. February 22, 2008. http://www.nytimes.com/2008/02/22/business/22device.html?scp=1&sq=Medical%20device%20ruling%20redraws%20lines%20on%20lawsuits&st=cse.16. Kessler DA, Vladeck DC. A critical examination of the FDA’s efforts to preempt failure-to-warn claims. Georgetown Law J. 2008;96:461-495.
17. Wright v Aventis Pasteur, PA Super 203 (2006); 905 A2d 544; Pa Super LEXIS 1728 (Pa Super. Ct 2006); Appeal denied, 591 Pa 674; 916 A2d 1103 (Pa 2007); dismissal entered, 2008 Phila Ct Com Pl LEXIS 221 (August 27, 2008).18. FDA embarks on Major hiring initiative for its public health mission [news release]. Rockville, MD: Food and Drug Administration; 2008. http://www.fda.gov/bbs/topics/NEWS/2008/NEW01829.html. Accessed September 29, 2008. 360k(a) (1976).14. Federal Food, Drug, and Cosmetic Act, 21 USC 301 et seq.15. Feder BJ. Medical device ruling redraws lines on lawsuits. New York Times. February 22, 2008. http://www.nytimes.com/2008/02/22/business/22device.html?scp=1&sq=Medical%20device%20ruling%20redraws%20lines%20on%20lawsuits&st=cse.16. Kessler DA, Vladeck DC. A critical examination of the FDA’s efforts to preempt failure-to-warn claims. Georgetown Law J. 2008;96:461-495.
17. Wright v Aventis Pasteur, PA Super 203 (2006); 905 A2d 544; Pa Super LEXIS 1728 (Pa Super. Ct 2006); Appeal denied, 591 Pa 674; 916 A2d 1103 (Pa 2007); dismissal entered, 2008 Phila Ct Com Pl LEXIS 221 (August 27, 2008).18. FDA embarks on Major hiring initiative for its public health mission [news release]. Rockville, MD: Food and Drug Administration; 2008. http://www.fda.gov/bbs/topics/NEWS/2008/NEW01829.html. Accessed September 29, 2008.
ARTICLES EN RAPPORT
Cette semaine dans le JAMA-Français
JAMA. 2008;300:1845.
Texte Complet
Actions régulatrices pour la sécurité des produits biologiques approuvés aux Etats-Unis et dans l’Union européenne
Thijs J. Giezen, Aukje K. Mantel-Teeuwisse, Sabine M. J. M. Straus, Huub Schellekens, et Antoine C. G. Egberts
JAMA. 2008;300:1887-1896.
Résumé
| Texte Complet
|

