|
|
PAGES DU PRATICIEN
Les lipoprotéines de haute densité en tant que cible thérapeutiqueUne revue systématique
Inder M. Singh, MD, MS;
Mehdi H. Shishehbor, DO, MPH;
Benjamin J. Ansell, MD
RÉSUMÉ
| |
Contexte Le cholestérol associé aux
lipoprotéines de haute densité (HDL-C) est un facteur de risque
cardiovasculaire dont l'intérêt en tant que cible
thérapeutique ne cesse de croître.
Objectifs Passer en revue les techniques, établies ou
émergentes, qui visent à modifier les lipoprotéines de
haute densité (HDL).
Sources de données Recherche systématique de la
littérature de langue anglaise, (1962 à mai 2007) sur les bases
de données MEDLINE et Cochrane, avec les mots-clés HDL-C et
apolipoprotein A-1 ; ainsi que les descripteurs : reverse cholesterol
transport, CVD [cardiovascular disease] prevention and control, drug therapy,
et therapy ; analyses des présentations réalisées dans
les rencontres cardiovasculaires majeures, entre 2003 et 2007 ; analyses
d'essais en cours, disponibles sur
ClinicalTrials.gov,
et des directives en vigueur publiées par les principales
sociétés cardiovasculaires.
Sélection des études et extraction des données
La priorité des études a été, dans l'ordre de
séquence, les essais randomisés, contrôlés, les
méta-analyses, et les études mécanistiques. Les
études de validation de principe et les essais de phase 1 à 3 de
nouvelles substances pharmacologiques ont également été
pris en compte. Deux auteurs ont évalué
l'éligibilité à l'étude, arbitré par un
troisième en cas de désaccord.
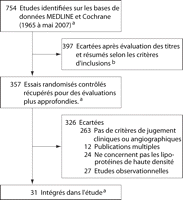
Synthèse des données Sur 754 études
identifiées, 31 essais randomisés contrôlés ont
rempli les critères d'inclusion. Les stratégies actuelles sur le
traitement ou le mode de vie, accroissent les niveaux de C-HDL de 20% à
30% lorsqu'elles sont optimisées. Alors que les études
préliminaires ont été encourageantes, les preuves de
l'effet bénéfique de l'accroissement du niveau de HDL-C sur la
prévalence des évènements cardiovasculaires,
indépendamment des variations des niveaux de lipoprotéines de
basse densité et de triglycérides, se sont montrées peu
tangibles. Des études menées chez l'homme montrent que des
nouveaux médicaments peuvent efficacement accroître le niveau de
HDL-C, alors que des nouvelles approches ciblant ses voies métaboliques
ou ses fonctions peuvent montrer très peu de résultats.
Conclusions Peu d'évidences supportent à ce jour un
accroissement massif du niveau de C-HDL par rapport à ce qu'il est
possible d'atteindre avec un changement du mode de vie. Les essais cliniques
en cours, ciblant des points spécifiques de la vois métabolique
des HDL pourraient offrir d'autres perspectives pour le traitement des
maladies cardiovasculaires.
JAMA.
2007;298(7):786-798
En dépit des avancées thérapeutiques, les
maladies cardiovasculaires (MCV) restent les principales causes de
mortalité et de morbidité dans les pays
développés. Bien que les statines réduisent les
coronaropathies d'environ 30%, un risque résiduel important subsiste,
même avec une diminution agressive du niveau de lipoprotéines de
basse densité (C-LDL). C'est pourquoi l'attention s'est aujourd'hui
déplacée sur des thérapies adjuvantes, ciblant les
lipoprotéines de haute densité (HDL) pour le traitement et la
prévention des MCV.
Nous passons en revue les essais randomisés contrôlés
publiés, portant sur l'influence des niveaux de cholestérol
lié aux lipoprotéines de haute densité sur les
résultats cliniques et l'imagerie d'athérosclérose. Nous
abordons également les observations épidémiologiques, les
études de validation de principe et mécanistiques, et les essais
majeurs de phase I à III concernant les nouveaux médicaments.
Finalement, nous évoquons les directives existantes et les nouvelles
approches encourageantes basées sur le HDL et apportons des
propositions d'applications cliniques.
ACQUISITION DES DONNEES
Avec l'aide d'un documentaliste médical, nous avons
réalisé une recherche systématique des
littératures de langue anglaise, de 1965 à mai 2007, sur les
bases de données MEDLINE et Cochrane, avec les mots-clés HDL-C
et apolipoprotein A-l, et les descripteurs reverse cholesterol transport, CVD
prevention and control, drug therapy, et therapy. Sept-cent cinquante quatre
études, dont une après mai 2007, ont été
identifiées; parmi lesquels 357 étaient des essais
randomisés contrôlés
(Figure 1). Nous avons
parcourus tous les résumés de ces études, analysant
l'ensemble de l'article dans de nombreux cas. Les recherches ont
été étendues aux références
bibliographiques des rapports de données et des articles de
synthèses les plus pertinents. Deux auteurs (I.M.S et M.H.S) ont
évalué indépendamment l'éligibilité de
chaque étude. En cas de divergence, l'ensemble du texte était
analysé avec un troisième auteur (B.J.A) et un consensus
atteint. La priorité que nous avons attribuée aux études
a été, dans l'ordre, les essais randomisés
contrôlés, les méta-analyses, et les études
mécanistiques. Les critères d'inclusion étaient (1)
d'être des essais randomisés portant sur l'accroissement des
niveaux de C-HDL ou la modification de la composition du HDL par des agents
pharmacologiques; (2) une affectation aléatoire aux traitements, et (3)
des critères cliniques ou de substitution (degré
d'athérosclérose) comme critère de jugement. Un total de
31 études a répondu aux critères
(Figures 1).
|
|
|
|
Figure 1.. Processus de sélection pour l'inclusion à l'étude
Voir « Acquisition des données » pour les termes de
recherche et les détails du processus de sélection.
a Inclut 1 étude identifiée après la date
d'exclusion, mai
2007.4
b Critères d'inclusion 1 et 2 (voir "Acquisition de
données").
|
|
|
Notre article inclut également des études
mécanistiques de bases et de validations de principes, ainsi que des
essais précliniques décisifs sur de nouveaux médicaments.
Par ailleurs, nous avons analysé les études observationnelles et
les méta-analyses, entre 1965 à 2007, et les
résumés présentés aux sessions annuelles de
l'American Heart Association et l'American College of Cardiology, de 2003
à 2007. Nous avons également analysé les directives de
l'American College of Cardiology, l'American Heart Association, du National
Cholesterol Education Program, de l'European Consensus Panel, et de l'American
Diabetes Association, ainsi que des essais de médicaments actuellement
en cours, sur
ClinicalTrials.gov.
SYNTHESE DES DONNEES
Corrélation entre le C-HDL et les MCV
Les études montrent qu'un niveau faible de C-HDL est fréquent
dans la population générale; 16% à 18 % des hommes, et 3%
à 6% des femmes5 ont un niveau inférieur à 35 mg/dl
(multipliez par 0.0259 pour convertir en mmol/l). D'autre part, un niveau
faible de HDL-C est un composant du syndrome métabolique, dont la
prévalence est de 24% chez les américains de plus de
20ans.6,7
De nombreuses études épidémiologiques ont
montré qu'un niveau faible de HDL-C était un facteur de risque
indépendant de
MCV.5,8
Dans la Framingham Heart Study par exemple, 43% à 44% des
évènements coronariens se sont produits chez des individus avec
des nivaux de C-HDL inférieurs à 40mg/dl (22% de la population
d'étude
totale).5 Les
individus ayant des niveaux de C-HDL inférieurs à 35mg/dl
avaient des incidences de MCV huit fois supérieures à ceux qui
avaient des niveaux supérieurs à
65mg/dl.5,8
La corrélation entre niveaux faibles de C-HDL et le risque accru de MCV
est également significative chez les individus âgés, et
pourrait être plus forte chez les
femmes.5,8,9
Les données angiographiques et échographiques indiquent que
les niveaux faibles de HDL-C sont associés au risque et à la
sévérité des coronaropathies, des maladies carotidiennes,
et de la resténose
post-angioplastie.9,10
Des études observationnelles ont montré que chaque baisse de 1
mg/dL en HDL-C plasmatique était associée à une
majoration de risque de MCV de 2% à 3%.9-10 D'autre part, chaque
augmentation de 1mg/dL était associée à une diminution du
risque de mortalité coronaire de 6%, indépendamment du taux de
C-LDL.9,10
Mécanismes de l'effet protecteur du HDL
Malgré l'effet protecteur contre les MCV attribué aux HDL,
les mécanismes par lesquels elles exercent leurs effets
antiathérogéniques sont au stade de la définition.
Plusieurs voies semblent impliquées, y compris le transport inverse du
cholestérol ainsi que des mécanismes qui ne dépendent pas
du
cholestérol.11
Transport inverse du cholestérol. Il s'agit du transfert du
cholestérol excédentaire des macrophages chargés en
lipides (cellules spumeuses), présents dans les tissus
périphériques, vers le foie, en faisant appel aux HDL,
aboutissant au catabolisme du cholestérol ou de son excrétion
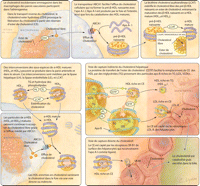
dans la bile (Figure
2).12
A l'intérieur de la paroi vasculaire, l'ester de cholestérol
emmagasiné dans les macrophages peut être transformé en
cholestérol libre par le cholestérol ester hydrolase, tandis que
l'acyl-coA cholestérol acyltransférase peut estérifier le
cholestérol cytosolique des macrophages qui se transforment en cellules
spumeuses athérogéniques.
|
|
|
|
Figure 2.. Vue schématique du transport inverse du cholestérol et du
métabolisme des lipoprotéines de haute densité
ABCA1: adenosine triphosphate-binding cassette transporter A1; ABCG1:
adenosine triphosphate-binding cassette transporter G1; apo:
apolipoprotéine; HDL: lipoprotéine de haute densité; LDL:
lipoprotéine de basse densité; LDL-R, Récepteur du LDL;
LPL: lipoprotéine lipase; SR-B1 scavenger receptor type B1; VLDL
lipoprotéine de très basse densité
|
|
|
Le devenir de HDL matures se décide entre au moins deux voies
métaboliques. Dans la voie directe, les cholestéryl esters
contenus au sein de la HDL peuvent être captés de façon
sélective par les hépatocytes et les cellules
sécrétrices de stéroïdes via un récepteur
éboueur de type B1 puis finissent excrété dans la
bile.12,14
Dans la voie indirecte, les cholestéryl esters peuvent être
échangées avec les triglycérides des particules riches en
apolipoprotéines B (LDL et lipoprotéines de très faible
densité [VLDL]) par l'action de la cholesteryl ester trasferase protein
(CETP). La captation des apolipoprotéines B riches en
cholestéryl esters par les récepteurs hépatiques du LDL
par la suite pourrait être responsable d'au moins 50% de transport
inverse du
cholestérol.12
Le HDL riche en triglycérides peut ensuite subir une hydrolyse par la
lipase hépatique et la lipase endothéliale pour donner des
petites HDL qui vont participer plus loin dans le
transport.12
Le rein semble jouer un rôle peu élucidé dans le
catabolisme des HDL, en contrôlant la vitesse de synthèse des apo
A-I pauvres en lipides.
Mécanismes non dépendant du cholestérol En plus de son
rôle majeur dans le transport inverse, la HDL possède d'autres
activités biologiques qui pourraient lui conférer ses effets
athéroprotecteurs.9,11,15
Celles-ci incluent des effets antioxydants (contrecarrant l'oxydation des
LDL), anti-inflammatoires, anti-thrombotiques/fibrinolytiques (diminuant
l'agrégation plaquettaire et la coagulation), et vasoprotecteur
(facilitant la décontraction des vaisseaux et inhibant le
chimiotactisme et l'adhésion des
leucocytes).9,11,1516
De manière collective, la HDL et ses constituants (y compris l'apo
A-I, la paraoxanase, le platelet activating factor acetyl-hydrolase, et
d'autres enzymes antioxydantes) exercent un ensemble d'effets qui pourraient
aider à prévenir l'athérosclérose, les syndromes
coronariens aigus, et la resténose après une angioplastie
coronaire.9,17
Hétérogénéité fonctionnelle des HDL
Les lipoprotéines de haute densité peuvent varier
considérablement en taille, densité, composition, et en termes
de propriétés fonctionnelles, ce qui définit probablement
leurs positions vis à vis de
l'athérogenèse.11,14,15,18,19
D'autre part, les niveaux de HDL-C plasmatiques ne permettent pas de
prédire ses rôles fonctionnels.
Les inflammations aigües ou chroniques peuvent avoir des
répercussions inattendues sur le rôle normalement protecteur des
HDL. Par exemple, la modification des apo A-1 au sein des HDL par la
myélopéroxidase leucocytaire altère la fonction des HDL
et leur donne des propriétés proinflammatoires et
athérogéniques.20
Les patients avec des MCV ou des maladies équivalentes ont des taux de
HDL proinflammatoires élevés par rapport à des sujets
sains. De plus, les propriétés inflammatoires et
anti-inflammatoires des HDL sont de meilleurs prédicteurs de la
prévalence de MCV que la HDL
seule.21
Dans la population de l'étude VA-HIL (Vet¬erans Affairs
High-Density Lipoprotein Cholesterol Intervention Trial), les particules HDL
 -1, riches en cholestérol constituaient le facteur de risque
principal pour les évènements cardiovasculaires
récurrents.21
Rader23
avait montré que 75% des patients ayant une maladie coronarienne ou des
maladies équivalentes avaient des HDL proinflammatoires malgré
un traitement de statines. Malgré l'abondance des techniques pour
séparer les HDL: par ultracentrifugation par gradient de
densité, résonance magnétique nucléaire, ou
électrophorèse sur gel à deux dimensions, il n'existe
aucune étude comparant la capacité de ces techniques à
prédire les évènements cardiovasculaires. De plus, les
étude mécanistiques divergent sur l'importance de la taille de
la particule dans la fonction des HDL, certaines montrant les particules de
petites tailles comme anti-inflammatoires, tandis que les autres mettent en
question le rôle protecteur de ces mêmes particules dans les
situations de stress oxydatifs
accrus.16,19-21 -1, riches en cholestérol constituaient le facteur de risque
principal pour les évènements cardiovasculaires
récurrents.21
Rader23
avait montré que 75% des patients ayant une maladie coronarienne ou des
maladies équivalentes avaient des HDL proinflammatoires malgré
un traitement de statines. Malgré l'abondance des techniques pour
séparer les HDL: par ultracentrifugation par gradient de
densité, résonance magnétique nucléaire, ou
électrophorèse sur gel à deux dimensions, il n'existe
aucune étude comparant la capacité de ces techniques à
prédire les évènements cardiovasculaires. De plus, les
étude mécanistiques divergent sur l'importance de la taille de
la particule dans la fonction des HDL, certaines montrant les particules de
petites tailles comme anti-inflammatoires, tandis que les autres mettent en
question le rôle protecteur de ces mêmes particules dans les
situations de stress oxydatifs
accrus.16,19-21
Les apolipoprotéines au sein des HDL sont des déterminants
clés de leurs fonctions. La principale apolipopotéine des HDL,
l'apo A-1, favorise la stimulation des activités ABCA1 et
lécithine-cholesterol acyl-transférase, et est un ligand du
récepteur éboueur de type B1. L'apo A-II, un autre constituant
des HDL a été montré comme
pro-athérogénique dans des modèles animaux. Les
stratégies thérapeutiques qui accroissent sélectivement
les niveaux d'apo A-I pourraient donc avoir plus d'effet
athéroprotecteur que celles qui modifient les taux d'apo A-I et d'apo
A-II à la fois.
Traitements non-pharmacologiques visant à accroître les taux de HDL-C
L'entraînement aérobie Il a été
montré que la pratique fréquente d'exercices aérobiques
accroît les niveaux de HDL-C d'environ 5% sur des périodes aussi
courtes que 2 mois, chez des individus sédentaires mais sains, à
travers plusieurs mécanismes
(Tableau
1).24,25
Pour obtenir un accroissement optimal des taux de HDL-C, les individus doivent
accomplir 5 exercices aérobie soutenues de 30 minutes par semaine une
durée totale cumulée; supérieure à 120 minutes par
semaine ayant le plus grand impact sur l'accroissement des niveaux de
HDL-C.42,43
|
|
|
|
Tableau 1.. Répercussions du changement de mode de vie sur les taux de C-HDL et
la composition en HDL
Abréviations: ABCA1, adenosine triphosphate-binding cassette
transporter A1; apo A-l, apolipoprotéine A-l; CETP cholestérol
ester transfer protéin; HDL-C, cholestérol associé au
lipoprotéines de haute densité; LCAT,
lécithine-cholestérol acyltransférase; LDL-C,
cholestérol associé au lipoprotéines de basse
densité; LPL, lipoprotéine lipase; AGMI, acide gras
mono-insaturé; AGPI, acide gras polyinsaturé.
Facteur de conversion SI: pour avoir HDL-C en mmol/l, multiplier par
0.0259
|
|
|
Le sevrage tabagique Le tabac, fumé ou on, abaisse les
niveaux de HDL-C (Tableau 1).
Une étude a montré que les niveaux de HDL-C augmentaient
d'environ 4mg/dL suivant un sevrage tabagique, sans être
accompagné de changements des niveaux de LDL-C, cholestérol
total, ou
triglycérides.27
La fumée de tabac est également une source de stress oxydatif
qui peut provoquer une altération fonctionnelle des HDL. Le sevrage
tabagique doit être fortement soutenu à travers une approche
multidisciplinaire, combinant l'accompagnement et méthodes
pharmacologiques si
nécessaire.42
La réduction pondérale La réduction
pondérale amène généralement à un
accroissement des niveaux de HDL-C (Tableau
1) chez les patients en surcharge pondérale ou
obèses.28 Pendant la baisse active du poids, les niveaux de HDL-C
diminuent légèrement, mais une fois la stabilité
atteinte, ils augmentent de 0.35mg/dL par kilogramme
perdu.29
Elle a été associée à une amélioration du
profil de risque cardio-métabolique lorsqu'elle est atteinte à
travers une modification du mode de vie, avec ou sans assistance
médicamenteuse.30-32
Les individus en surcharge pondérale ou obèses devraient avoir
comme objectif un indice de masse corporelle (calculé comme le poids en
kilogramme divisé par le carré de la taille en mètre)
inférieur à 25, avec une baisse de 2 kg par
mois.42
Consommation d'alcool L'absorption de quantités
modérées d'alcool (30-40g [1-3 verres] par jour) accroit les
niveaux de HDL-C et est associée à une diminution du risque de
maladie coronarienne, indépendamment des autres
facteurs.33,34
Cette corrélation est probablement derrière le
phénomène "sick quitter" où des consommateurs
réguliers d'alcool arrêtent leurs consommations à cause de
la maladie ou sur conseil médical et sont classés à tort
comme
non-consommateurs.35
L'ingestion de 30 à 40 g d'alcool par jour pendant 3 semaines permet
d'avoir des accroissements des niveaux de HDL-C allant jusqu'à 12%,
quel que soit le type d'alcool consommé
(Tableau
1).34
Cependant, les recommandations actuelles se limitent à 2 verres par
jour chez les hommes et à un verre chez les
femmes.42
Les personnes non-consommatrices ne doivent pas être conseillées
à consommer régulièrement de l'alcool.
Facteurs alimentaires Des régimes riches en acides gras
saturés et en acides gras trans peuvent induire un accroissement des
niveaux de HDL-C, mais également de LDL-C et de l'expression des
molécules d'adhésion des cellules endothéliales, induite
par les
HDL.36 En
revanche, les régimes riches en acides gras polyinsaturés
améliorent la capacité anti-inflammatoire des
HDL.36 Le
remplacement des acides gras saturés et acide gras trans par des acides
gras monoinsaturés et des acides gras polyinsaturés induit une
diminution du rapport
LDL-C/HDL-C.37
L'absorption d'acides gras polyinsaturés en n-3 a été
associée à un accroissement des niveaux de HDL-C et à des
bénéfices
cardiovasculaires38;
cependant, les études sur les effets cardiovasculaires des acides gras
polyinsaturés en n-6 font
défaut.39
Le remplacement des acides gras saturés et des acides gras trans par
des hydrates de carbone à faible index glycémique pourrait
améliorer le profil HDL-C mais les données sont
insuffisantes.40
Des données récentes montrent qu'un régime à
faible teneur en graisse, et élevé en fibres, pendant 2 à
3 semaines, associé à de l'exercice physique, font passer les
HDL d'un état proinflammatoire à un état
anti-inflammatoire.25
Cette amélioration de la propriété fonctionnelle
s'observait malgré une faible réduction des taux de HDL-C,
suggérant une intensification du recyclage des HDL
proinflammatoires.25
Le remplacement des aliments riches en acides gras saturés et acides
gras trans par des sources en acides gras monoinsaturés et
polyinsaturés comme les huiles végétales (olive, soja,
moutarde, colza, lin), les noix (amandes, arachides, noisettes, pécan),
et les fruits de mer (saumon, thon, maquereau, huiles marines) devrait
être recommandé aux
patients.42
Dans leur majorité, les études sur les changement du mode de
vie ont montré des bénéfices vis à vis des MCV;
cependant un seul mécanisme ou une seule stratégie ne suffirait
pas à rendre compte de
ceux-ci.30,41
Médicaments disponibles pour accroître les niveaux de HDL-C
Les traitements médicamenteux actuels à base de HDL peuvent
être regroupés en 2 catégories principales: les
médicaments qui accroissent ou réduisent les taux des
constituants de l'HDL (HDL-C, apo A-I, phospholipides), et les
médicaments qui exercent une régulation positive sur le
transport inverse du cholestérol et de l'efflux de cholestérol
des macrophages.
Médicaments modifiant la composition des HDL L'acide nicotinique Mis
en évidence à l'origine comme un traitement de la
dyslipidémie, l'acide nicotinique permet d'accroitre le niveau de HDL-C
de 20% à 30%, ainsi que de réduire le niveau de
triglycérides plasmatiques de 40% à 50% et le niveau de LDL-C
jusqu'à 20% (Tableau
2).44-56
C'est le médicament le plus efficace pour augmenter les taux de HDL-C
à l'heure actuelle, lorsqu'il est
toléré.9
Les preuves les plus fortes en faveur de l'utilisation de la niacine
proviennent de l'étude Coronary Drug Project (CDP) qui a
évalué la monothérapie à la niacine chez 8 341
hommes avec un antécédent d'infarctus du myocarde. Dans cette
étude avant traitement à la statine, la niacine a
été associée à une réduction de 27% de
l'incidence des réinfarctus non-fatals à 6 ans, et une
réduction de la mortalité globale à 15 ans de 11%
(Tableau
3).50
|
|
|
|
Table 2.. Effets des traitements médicamenteux actuellement disponibles sur
les taux de HDL-C et la composition en HDL.
Abréviations: ABCG1, adenosine triphosphate-binding cassette
transporter G1; apo A-l, apolipoprotéine A-l; apoC-lll,
apolipoprotéine C-lll; ALT, alanine aminotransférase; AST,
aspartate aminotransférase; CETP, cholestérol ester transfer
protein; ICC, insuffisance cardiaque congestive; SNC, système nerveux
central; CPK, créatinine phosphokinase; MCV, maladies
cardiovasculaires; DGAT, diacylglycerol acyltransférase; LM,
libération modifiée; HDL-C, cholestérol associé au
lipoprotéines de haute densité; LDH, lactate
déshydrogénase; LPL, lipoprotéine lipase; ARNm, acide
ribonucléique messager; PPAR, peroxisome proliferator-activated
receptor; LP, libération prolongée; IVRS, infection de la voie
respiratoire supérieure.
a Les bouffées congestives peuvent être
minimisées par une posologie à dose croissante de LM niacine
(commencez à 500mg par nuit et augmenter de 500mg par mois
jusqu'à atteindre 1g à 2g par nuit), une prise d'aspirine une
demi-heure avant, en évitant les aliments/boissons chauds,
épicés avant; manger un en-cas pauvre en graisse.
b Disponible uniquement en dehors des Etats-Unis à
l'heure actuelle
|
|
|
|
|
|
|
Table 3.. Essais randomisés contrôlés de traitements
médicamenteux modifiant HDL et impactant les résultats cliniques
ou le degré d'athérosclérose en critère de
substitution
Abréviations: AFREGS, Armed Forces Regression Study; apo A-1,
apolipoprotéine A-l; ARBITER, Arterial Biology for the Investigation of
the Treatment Effects of Reducing Cholesterol; BECAIT, Bezafibrate Coronary
Atherosclerosis Intervention Trial; BIP, Bezafibrate Infarction Prevention
Study; CP coronaropathie ; CDP, Coronary Drug Project; CLAS, Cholesterol
Lowering Atherosclerosis Study; CLAS Fern, groupe CLAS de
l'athérosclérose fémorale; CLAS IMT, groupe CLAS de la
carotide à l'ultrason; DAIS, Diabetes Atherosclerosis Intervention
Study; ERASE, Effect of rHDL on Atherosclerosis-Safety and Efficacy Trial;
FATS, Familial Atherosclerosis Treatment Study; FIELD, Fenofibrate
Intervention and Event Lowering in Diabetes Study; HATS, HDL-Atherosclerosis
Treatment Study; HDL-C, cholestérol associé aux
lipoprotéines de haute densité; HHS, Helsinki Heart Study;
ILLUSTRATE. Investigation of Lipid Level Management Using Coronary Ultrasound
to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation
Trial; EIM, épaisseur de l'intima-media; IVUS, échographie
intravasculaire; LEADER, Lower Extremity Arterial Disease Event Reduction
Trial; LOCAT, Lopid Coronary Angiography Trial; MI, infarctus du myocarde; NR
non rapporté; RADIANCE, Rating Atherosclerotic Disease Change by
Imaging with a New CETP Inhibitor Trial; SCRIP, Stanford Coronary Risk
Intervention Project; AIT, attaque ischémique transitoire; VA-HIT,
Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial; WHO,
World Health Organization.; ACV attaque cérébrovasculaire
a Le décès correspond à la mortalité
globale.
|
|
|
D'autres études des résultats cliniques de la niacine ont
été menées dans le cadre d'une thérapie
combinée et les résultats ainsi obtenus doivent être
interprétés dans ce contexte. Globalement, les traitements
à la niacine, en monothérapie ou combinée à
d'autres médicaments anti-dyslipidémiques, se sont traduits par
des bénéfices significatifs sur le plan clinique et le
degré d'athérosclérose
(Tableau 3). La plupart des
bénéfices cardiovasculaires sont attribuables à ses
propriétés à accroître le niveau de HDL-C
(Tableau
2).9
Le potentiel thérapeutique de la niacine est limité par ses
effets secondaires, qui peuvent être atténués par une
administration à libération prolongée. L'American
Diabetes Association met en garde contre son utilisation chez les patients
diabétiques, bien que de récentes études n'aient
montré aucune augmentation significative de la glycémie à
long terme, des traitements hyperglycémiques oraux, ou d'interruption
de la niacine chez ces
patients.85,86
Statines Les statines entraînent une augmentation modérée
du niveau de HDL-C de 5% à 15%, l'effet le plus notable se manifestant
avec la
rosuvastatine.68
Le mécanisme par laquelle les statines induisent l'augmentation des
HDL-C est peu compris, mais elles entraînent l'accroissement des niveaux
d'apo A-I et de HDL pauvres en lipides, ainsi que l'activité de la
paraoxonase, une enzyme anti-oxydante
(Tableau
2).69-72
Dans une analyse post hoc récente de 1455 patients ayant de faibles
taux de HDL-C, les statines ont, indépendamment des LDL-C,
réduit les athéromes coronariens lorsque les taux de HDL-C
augmentaient d'au moins 7.5%. Elles entraînent une diminution plus
importante des MCV chez les patients ayant des niveaux de HDL-C plus faibles,
indépendamment des effets des LDL-C. Cependant, du fait de leurs effets
peu importants sur le niveau de HDL-C, elles ne sont
généralement pas adaptées pour une monothérapie
visant à les
accroître.75
Médicaments ciblant la voie de transport inverse du
cholestérol et l'efflux de cholestérol des macrophages Les
dérivés de l'acide fibrique (les fibrates), qui sont des
agonistes du récepteur activé par la
prolifération des peroxysomes (PPAR), peuvent accroître le niveau
de HDL-C (de 10% à 20%), abaisser faiblement le LDL-C (10% à
15%), et abaisser significativement le niveau de triglycérides (40%
à 50%) (Tableau
2).57,58
Plusieurs études ont élucidé les effets des fibrates sur
le transport inverse du cholestérol
(Tableau
2).87,88
Dans la Helsinki Heart Study et l'essai VA-HIT, le traitement au
gemfibrozil a été associé à une réduction
des évènements cardiovasculaires
(Tableau 3). La
réduction de 22% des évènements coronariens dans le
VA-HIT a été attribuée à une augmentation modeste
(6%) du niveau de HDL-C. A contrario, les résultats des études
sur les fibrates comme le bézafibrate et le fénofibrate ont
été négatifs tandis que le clofibrate a été
associé à un risque potentiel
(Tableau
3).44,57-67,80
Des indications tendent à attribuer les bénéfices
observés dans le VA-HIT aux effets additifs des fibrates sur la
synthèse des apolipoprotéines et le métabolisme des
lipoprotéines (Tableau
2).89-90
La combinaison de fibrates, en particulier le gemfibrozil, avec des
statines nécessite des précautions et une surveillance des taux
de créatine kinase du fait du risque de myotoxicité, y compris
de
rhabdomyolyse.42
Au regard de ce risque et des résultats variables observés, le
positionnement du traitement aux fibrates reste incertain mais inclut
vraisemblablement les patients exposés à des risques
élevés de coronaropathie, de faibles niveaux de HDL-C, et des
niveaux élevés de triglycérides sériques.
Les effets de l'ajout du fénofibrate à un traitement de
statines sur le plan cardiovasculaire fait actuellement l'objet d'un essai
clinique à grande échelle chez des patients diabétiques
de type
2.91
Nouveaux traitements à base de HDL
Plusieurs nouvelles approches thérapeutiques basées sur le
HDL ont été ou sont actuellement évaluées dans des
essais cliniques de phase I à III. Celles-ci incluent des nouvelles
formulations des thérapies basées sur l'acide
nicotinique/agonistes du récepteur à l'acide nicotinique, les
inhibiteurs de la CETP, les antagonistes du récepteur cannabinoïde
type 1, les agonistes du PPAR, les agonistes du récepteur X/farnesoid X
du foie, et les apo A-I et/ou les phospholipides
(Tableau 4). A l'instar des
thérapies actuelles, les nouvelles approches, en stade
d'évolution, peuvent également être classées en
tant que médicaments accroissant ou modifiant les taux des constituants
des HDL et en tant que médicaments régulant positivement le
transport inverse du cholestérol et l'efflux de cholestérol des
macrophages.
|
|
|
|
Table 4.. Effets des stratégies pharmacologiques prometteurs sur le niveau de
HDL-C et les constituants de la HDL
Abréviations: ABCA1, adenosine triphosphate-binding cassette
transporter A1; ABCGt, adenosine triphosphate-binding cassette transporter G1;
apo A-l, apolipoprotéine A-l CETP, cholesterol ester transfer protein;
DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphocholine;HDL-C, cholestérol
associé aux lipoprotéines de haute densité; GLU, gros
liposomes unilamellaire NA, non applicable car variations du niveau de HDL-C
non mesurées; rApo A-l, apolipoprotéine A-l recombinante; PPAR,
peroxisome proliferator-activated receptor.
3MK-0524 est actuellement en évaluation sous deux formes
combinées: MK-0524A (antagoniste sélectif du récepteur 1
de la prostaglandine D2 + niacine à libération modifiée)
et MK-0524B (antagoniste sélectif du récepteur 1 de la
prostaglandine D2 + niacine à libération modifiée +
simvastatine)
|
|
|
Médicaments en développement modifiant la composition des HDL
Formules basées sur l'acide nicotinique Il a été
démontré récemment que le MK-0524, un antagoniste
sélectif du récepteur 1 de la prostaglandine réduisait
les vasodilatations et les bouffées congestives associées
à l'acide nicotinique chez la souris et chez l'homme
(Tableau
4).92
L'effet du MK-0524 et de la niacine à libération
prolongée sur les MCV est actuellement évalué dans un
essai sur des patients présentant une maladie coronarienne et de
faibles niveaux de HDL-C, et recevant un traitement de
statines.110
Inhibiteurs de la CETP. L'inhibition de la CETP se traduit par un
accroissement des taux de HDL-C et une réduction des taux de VDL, LDL-C
et de triglycérides (Tableau
4).93
La multiplication des particules larges et de densité faible de HDL est
également favorisée, ce qui peut conduire à un transport
inverse de cholestérol, indépendant des voies impliquant le
pré-?-HDL et
l'ABCA1.94
Les indications épidémiologiques sur l'effet du CETP sont
contradictoires,95-97
néanmoins l'accroissement notable des niveaux de HDL-C, observés
pour des composés en développement qui exhibent une forte
activité inhibitrice pour la CETP, a entraîné le
développement commercial de ces médicaments.
Il a été montré que le torcetrapib, un inhibiteur
direct de la CETP, réduisait l'athérosclérose aortique
chez l'animal et accroissait significativement les niveaux de HDL-C chez
l'homme. Le développement a été cependant interrompu
suite aux résultats de l'essai ILLUMINATE (Investigation of Lipid Level
Management to Understand Its Impact in Atherosclerotic Events) qui avait
montré une augmentation de 61% de la mortalité globale et autres
évènements cardiovasculaires chez les individus exposés
à un risque élevé de maladie coronarienne et
traités avec le torcetrapib plus atorvastine, par rapport au groupe
contrôle recevant un placebo avec atorvastine. Des analyses provenant
des essais sur le torcetrapib ont montré que son utilisation
était associée à une augmentation moyenne de 3 à 4
mm Hg de la pression sanguine, un phénomène qui semble
spécifique à la molécule et sans rapport avec son
activité
inhibitrice.114
Trois autres essais évaluant l'effet du composé sur
l'athérome coronaire par doppler intra-coronaire et mesure de
l'épaisseur intima-média de la carotide, n'ont montré
aucun changement quant au degré d'athérosclérose
malgré un accroissement marqué des taux de HDL-C
(Tableau
3).4,81,82
Bien que le mécanisme exact rendant compte des fréquences
plus élevées d'évènements indésirables dans
l'essai ILLUMINATE ne soit pas élucidé, les explications
probables incluent (1) l'accroissement de la pression sanguine et d'autres
effets hémodynamiques non recherchés. Dans ILLUSTRATE, une
augmentation moyenne de 4.6 mm Hg était observée pour le groupe
torcetrapib par rapport au groupe contrôle. Il s'agit d'une
toxicité spécifique au composé, dans la mesure où
les mêmes effets n'ont pas été rapportés pour
d'autres classes d'inhibiteurs de la CETP, ou chez des patients ou animaux
déficients pour cette molécule.98 (2) Production de HDL
dysfonctionnels, l'inhibition de la CETP conduit à la production de
particules -HDL, qui ne sont pas les transporteurs
privilégiés dans l'efflux du cholestérol
médié par l'ABCA1. (3) Production de HDL proinflammatoire,
probablement par oxydation des phospholipides des particules de HDL qui se
sont accumulées dans la cellule après l'inhibition de la CETP.
(4) Des interactions non prédites entre le torcetrapib et
l'atorvastatine. Les futures études sur des médicaments de la
même classe qui n'affectent pas la pression sanguine apporteront
peut-être la lumière sur le(s) mécanisme(s) mettant en
échec le
torcetrapib.114
Le JTT-705, un inhibiteur de la CETP, réduit l'activité de
celle-ci de 95% chez le lapin, avec une augmentation de 90% des niveaux de
HDL-C, une baisse de 40% des non-HDL-C et une baisse de 80% de
l'athérosclérose aortique
(Tableau 4).100 Les
données provenant de l'animal concernant l'effet du JTT-705 sur
l'athérosclérose sont cependant discordantes. Une étude
chez des lapins hypercholestérolémiques a montré que
même avec des modifications similaires du profil lipidique, le JTT-705
n'affectait par l'athérosclérose. Ces études
étaient potentiellement limitées par les taux
élevés de cholestérol total et de triglycérides,
qui peuvent contrecarrer les effets bénéfiques de
l'accroissement des taux de HDL-C.
Des essais de phase II récents, randomisés, en double
aveugle, et contrôlés par placebo, ont montré que JTT-705,
utilisé seul ou combiné avec de la pravastatine était
bien toléré et non associée à une
altération de la pression sanguine. Cependant, à ce jour, aucun
essai évaluant des résultats cliniques avec le JTT-705 n'ont
été menés à terme.
Traitements ciblant l'apo A-I Les stratégies actuelles basées
sur l'apo A-I incluent la perfusion de phospholipides recombinants
complexés à l'apo A-I (rHDL), de la proapoliprotéine A-I
recombinante, de l'apo A-I, ou des peptides mimétiques de l'apo A-I, ce
qui accroît la quantité circulante d'accepteurs du
cholestérol pauvres en lipides
(Tableau 4).
Il a été montré en doppler intravasculaire que les
perfusions hebdomadaires de rHDL avec l'ETC-216, un complexe recombinant
apoA-I Milano/phospholipides, favorisait la régression des plaques
d'athéromes coronaires après 5 semaines chez des patients ayant
des syndromes coronaires aigus (Tableau
3).83 Par ailleurs, l'essai ERASE (Effect of rHDL on
Atherosclerosis-Safety and Efficacy) a évalué les effets du rHDL
chez 183 patients ayant des syndromes coronaires aigus en utilisant le doppler
intravasculaire et les résultats angiographiques.84 Comparé au
placebo, le critère de jugement primaire qui était le
pourcentage de changement en volume de l'athérome, aucune
différence significative n'a été constatée
(Tableau
3).84
L'efflux de cholestérol des cellules spumeuses par la voie ABAC1 est un
des mécanismes anti-athérogéniques possibles dans ces
stratégies de
perfusion.83,84
Les mimétiques de l'apo A-I à 243 acides aminés sont
des petits peptides semblables au domaine de fixation lipidique de l'APo A-I
dont les effets athéroprotecteurs ont été montré
dans des modèles animaux.105 L'un de ces mimétiques, le L-4F, a
été montré in vivo pour améliorer la
vasodilatation et empêcher l'adhésion des monocytes, tandis que
le D-4E, un composé dérivé oral, qui est
protégé contre les enzymes digestives réduisait
significativement l'athérosclérose dans des modèles
animaux (Tableau
4).105
Le mécanisme expliquant ces effets serait la production rapide de
particules β migrantes, accompagnée d'un renforcement de
l'activité paraoxonase, de la capacité anti-inflammatoire, et de
la capacité à favoriser l'efflux de cholestérol des
macrophages.105,106,115
L'administration orale du D-4E favorisait le transport inverse du
cholestérol aussi bien in vitro qu'in vivo, en accélérant
les étapes initiales du transport
inverse.106
Dans un essai de phase I chez l'homme, une dose orale unique de D-4F
était associée à une amélioration de la fonction
HDL anti-inflammatoire. Les résultats d'une administration
parentérale du peptide ETC-642 sont attendus d'ici peu. Cependant, ces
traitements basés sur l'apo A-I n'entrainent pas un accroissement des
HDL-C mais génèrent plutôt des particules de type HDL-C,
modifie la composition des HDL, ou fait les deux à la fois.
Traitements ciblant les phospholipides Les phospholipides, un des
constituants des HDL, sont également des cibles thérapeutiques
(Tableau
4).107
Dans un modèle murin apoE null, le
1,2-dimyristoyl-sn-glycero-3-phosphocholine, un phospholipide
synthétique oral, avait permis d'obtenir un accroissement des taux de
HDL-C et d'apo A-I, accompagnée d'une amélioration des fonctions
HDL et une réduction de la taille des lésions
aortiques.107
Dans une petite étude (n=16) chez l'homme, du phosphatidyl inositol
oral, dérivé de la lécithine de soja, a montré un
accroissement des taux de HDL-C et d'apo A-I
(Tableau 4).108 Par ailleurs,
des perfusions de particules de phospholipides (phosphatidylcholine ou de gros
liposomes unilamellaires) ont été proposées pour
améliorer le transport inverse du cholestérol et la
régression de
l'athérosclérose.109
Bloqueurs des récepteurs cannabinoïdes de type 1 Il a
été montré que le rimonabant, premier bloqueur
sélectif des récepteurs cannabinoïdes de type 1
possédant des propriétés anorexigènes, augmentait
de manière dose-dépendante et indépendamment de la perte
de poids les taux de HDL-C de 10% (Tableau
2).31,32
Comparé à un placebo, les patients recevant 20 mg de rimonabant
avaient des taux de HDL-C deux fois supérieurs à ce qui est
attendu après une perte de poids uniquement; ce qui suggère un
effet pharmacologique direct du rimonabant sur le métabolisme
lipidique.31,32
De plus, les taux de HDL-C augmentaient de façon continue pendant les
deux années de l'étude, bien que la masse corporelle ait
été
stabilisée.31,32
Plusieurs essais à grande échelle évaluant l'effet du
rimonabant sur les résultats cliniques sont actuellement en cours;
toutefois le médicament n'a pas été approuvé aux
Etat-Unis pour cause de toxicité possible au niveau du système
nerveux central.
Médicaments en développement ciblant la voie de transport
inverse du cholestérol et l'efflux de cholestérol des
macrophages Les agonistes du PPAR dont les rôles dans l'accroissement du
taux de HDL-C sont actuellement à l'étude sont répartis
en 3 classes: PPAR-, PPAR-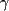 , et PPAPR- , et PPAPR-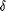 (précédemment appelé PPAR-β), appartenant toutes
à la famille des facteurs de transcription des récepteurs
nucléaires qui sont impliqués dans le métabolisme des
acides gras. Contrairement à la première
génération d'agonistes du PPAR- (fibrates), les nouvelles
molécules, comme le NS-20, sont plus sélectives et plus
puissantes (Tableau
4).103
La non-toxicité d'un de ces agonistes, le
LY18674, a
été mise en question, notamment sur l'accroissement des taux de
taux de créatinine et de LDL-C. Les activateurs du PPAR-
employé pour le diabète de type II, comme les
thiazolidinediones, accroissent les taux de HDL-C de 5% à 15%
(Tableau 2); cependant ils ont
été associés à une prévalence accrue des
MCV.78 Le
GW50516, un
agoniste du PPAR- accroit les taux de HDL-C de 80% dans le
modèle simien (Tableau
4).104
Les résultats d'un essai de phase II chez l'homme est toujours en
attente.117
(précédemment appelé PPAR-β), appartenant toutes
à la famille des facteurs de transcription des récepteurs
nucléaires qui sont impliqués dans le métabolisme des
acides gras. Contrairement à la première
génération d'agonistes du PPAR- (fibrates), les nouvelles
molécules, comme le NS-20, sont plus sélectives et plus
puissantes (Tableau
4).103
La non-toxicité d'un de ces agonistes, le
LY18674, a
été mise en question, notamment sur l'accroissement des taux de
taux de créatinine et de LDL-C. Les activateurs du PPAR-
employé pour le diabète de type II, comme les
thiazolidinediones, accroissent les taux de HDL-C de 5% à 15%
(Tableau 2); cependant ils ont
été associés à une prévalence accrue des
MCV.78 Le
GW50516, un
agoniste du PPAR- accroit les taux de HDL-C de 80% dans le
modèle simien (Tableau
4).104
Les résultats d'un essai de phase II chez l'homme est toujours en
attente.117
L'agonisme des PPAR a été proposé comme intensifiant
l'efflux de cholestérol des macrophages et des propriétés
anti-inflammatoires des HDL. Il a été montré que les
doubles agonistes du PPAR- et PPAR-, comme le muraglitazar
augmentaient le taux de HDL-C jusqu'à 16% ; cependant une analyse
d'essais de phase II et III de celle-ci a montré une augmentation
significative du critère d'évaluation composite comprenant
mortalité globale, infarctus du myocarde non fatal, accident
cérébro-vasculaire, accident ischémique transitoire, et
insuffisance cardiaque congestive ; entrainant l'arrêt de son
développement.
COMMENTAIRES
La corrélation inverse entre le taux de HDL-C et
l'athérosclérose est confortée par les enquêtes
cliniques et les explorations scientifiques de base. La HDL accroît le
transport inverse de cholestérol et possède des effets
antioxydants, anti-inflammatoires, antithrombotiques, et
vasprotecteurs.11,15,16
Malgré l'abondance de preuves associant les faibles niveaux de HDL-C
à la mortalité et la morbidité cardiovasculaire, il n'y a
pas d'indication pour conclure définitivement que l'augmentation des
taux de HDL-C réduit l'incidence des évènements
cardiovasculaires majeurs. Les principales directives ont de ce fait
évité de désigner le HDL-C comme cible malgré
certaines déclarations allant dans le sens du consensus
général.9,42
Les directives actuelles de l'Adult Treatment Panel III insistent sur le
fait de donner la priorité aux LDL-C, ensuite les non-HDL-C avant de
s'adresser aux HDL-C. La dernière déclaration scientifique de
l'American Heart Association/National Heart, Lung, and Blood Institute propose
le HDL-C en tant que "cible tertiaire" (après les LDL-C et
triglycérides), avec en objectif des niveaux de HDL-C supérieurs
à 40 mg/dL et 50mg/dL respectivement chez les hommes et les femmes.
L'American Diabetes Association propose le HDL-C en tant que "cible
secondaire" au même titre que les triglycérides, avec en
objectif des niveaux supérieurs à 40 mg/dL. Les deux organismes
consultatifs recommandent la niacine et les fibrates pour accroitre le niveau
de HDL-C si le changement de mode de vie se révèle
insuffisant.
Les nouvelles approches basées sur la HDL peuvent être
réparties entre celles qui modulent la composition de l'HDL-C (HDL-C,
apo A-I et phospholipides) et celles qui favorisent le transport inverse du
cholestérol. Une troisième catégorie, modulant les
fonctions anti-inflammatoires et antioxydantes des HDL se retrouve dans les
deux approches précédentes et pourrait être
développée indépendamment. Les échecs des
molécules récentes qui augmentent significativement les taux de
HDL-C suggèrent que les fonctions des HDL pourraient constituer une
meilleure cible que les molécules elles-mêmes. De plus, la
relation entre l'inflammation systémique et la fonction des HDL
pourrait revêtir un caractère essentiel.
Les fonctionnalités des différentes sous-fractions de HDL
semblent relativement hétérogènes. Parmi les formes
connues de l'HDL-C (pré-β-HDL, HDL2, HDL3), la
pré-β-HDL apparaît comme la plus
anti-athérogénique. De ce fait, les traitements visant à
augmenter cette sous-fraction devraient offrir le plus de promesses. Par
ailleurs, les évaluations fonctionnelles des HDL pourraient apporter
plus de clarté sur les promesses thérapeutiques des
médicaments à l'étude.
CONCLUSIONS
Si les stratégies visant à réduire les LDL-C
résultent souvent en une diminution du risque de MCV, les approches
basées sur les HDL doivent faire face à plus de
complexité et se révèlent parfois décevantes. Au
cours de l'année dernière, le développement du pactimibe,
un inhibiteur de l'acylcholestérol acyltransférase, et cette
année, celui du torcetrapib, ont été abandonnés.
Ces interruptions font suite à la mise en évidence d'effets
secondaires chez certains membres de la classe PPAR.
Les résultats négatifs obtenus avec ces composés ne
mettent pas en cause le concept consistant à accroitre le taux de
HDL-C, cibler les fonctions des HDL, ou les deux à la fois pour traiter
l'athérosclérose. Cependant, l'objectif, à priori simple,
d'accroitre le taux de "bon cholestérol" n'est plus
envisageable pour toutes les formes de HDL sans prendre en
considération les effets thérapeutiques sur la fonction des HDL
et à terme le risque cardiovasculaire. La corrélation entre
l'altération fonctionnelle des HDL et les résultats
d'études à long terme pourrait permettre de valider
prospectivement des procédures d'évaluation la fonction des
HDL.
En attendant, les stratégies appropriées pour accroître
les taux de HDL-C comprennent le changement de mode de vie (exercice physique,
régime, réduction pondérale, arrêt du tabac). Les
patients exposés à des risques élevés de MCV et
présentant de faibles taux de HDL-C peuvent bénéficier
d'un traitement à base de statines et de niacine, malgré l'effet
relativement modeste des statines sur le taux de HDL-C. La niacine et les
fibrates méritent également d'être pris en
considération, dans le cadre d'une monothérapie ou
combinées à un traitement à la statine et faisant l'objet
d'une étroite surveillance chez les patients exposés à un
risque élevé de maladies coronariennes.
Informations sur les auteurs
Correspondance: Mehdi H. Shishehbor, DO, MPH, Cleveland Clinic, 9500
Euclid Ave, JJ40, Cleveland, OH 44195
(shishem{at}gmail.com).
FMC disponible en ligne à
www.jama.com
Contributions de l'auteur: Le Dr Singh a eu un accès complet
à toutes les données de l'étude et accepte la
responsabilité de l'intégrité des données et de
l'exactitude de l'analyse des données.
Conception et schéma de l'étude : Singh, Shishehbor,
Ansell.
Recueil des données : Singh, Shishehbor.
Analyse et interprétation des données : Singh,
Shishehbor, Ansell.
Rédaction du manuscrit : Singh, Shishehbor, Ansell.
Revue critique du manuscrit : Singh, Shishehbor, Ansell.
Aide administrative, technique et matérielle : Singh,
Shishehbor, Ansell.
Supervision de l'étude : Shishehbor, Ansell.
Les Dr Singh et Shishehbor ont également contribué ce
manuscrit.
Liens financiers: Le Dr Ansell a rapporté avoir reçu
des honoraires d'orateurs d'AstraZeneca, Kos Pharmaceuticals, Merck, et
Pfizer; des traitements de recherche de Merck et Pfizer dans le passé;
et d'avoir des intérêts chez Bruin Pharma. Aucun autre lien
financier n'a été rapporté.
Financement/soutien: Le Dr Shishehbor a
bénéficié du soutien partiel du National Institutes of
Health, National Institute of Child Health and Human Development,
Multidisciplinary Clinical Research Career Development Programs bourse K12
HD049091 et du National Institutes of Health Loan Repayment Program. Le Dr
Ansell a bénéficié du soutien privé
philanthropique de Mr et Mrs Timothy Hannemann, Ms Kathleen Kennedy, Mr Frank
Marshall, Mr et Mrs Michael Ovitz, et Mr et Mrs Nelson Rising.
Rôle des sponsors: Aucun des Instituts Nationaux de
Santé ni les personnes ayant soutenu le Dr Ansell de façon
philanthropique n'ont joué un rôle dans le schéma et la
conduite de l'étude, le recueil, l'analyse et l'interprétation
des données; ni dans la préparation, la revue ou l'approbation
du manuscrit.
Autres Contributions: Alan M. Fogelman, MD (Department of Medicine
and Atherosclerosis Research Unit, University of California, Los Angeles), a
apporté des critiques utiles et pertinentes sur le manuscrit, et Dave
Schumick, BS, CMI (Center for Medical Art and Photography, Cleveland Clinic,
Cleveland, Ohio), a fourni son aide pour les images. Aucun n'a reçu de
compensation financière.
Affiliations des auteurs: Department of Cardiovascular Medicine,
Krannert Institute of Cardiology, Indiana University Medical Center,
Indianapolis; Department of Cardiovascular Medicine, Cleveland Clinic,
Cleveland, Ohio; and Department of Medicine, David Geffen School of Medicine
at UCLA, Los Angeles, California.
BIBLIOGRAPHIE
| |
1. Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate
lipid lowering with statins after acute coronary syndromes. N Engl
J Med.2004; 350(15):1495
-1504.2. Downs JR, Clearfield M, Weis S, et al; AFCAPS/TexCAPS Research
Group. Primary prevention of acute coronary events with lovastatin in men and
women with average cholesterol levels: results of AFCAPS/TexCAPS.
JAMA.1998; 279(20):1615
-1622.3. LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering
with atorvastatin in patients with stable coronary disease. N Engl
J Med.2005; 352(14):1425
-1435.4. Bots ML, Visseren FL, Evans GW, et al; RADI-ANCE 2 Investigators.
Torcetrapib and carotid intima-media thickness in mixed dyslipidaemia
(RADIANCE 2 study): a randomised, double-blind trial.
Lancet.2007; 370(9582):153
-160.5. Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S,
Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol
levels: the Framingham Study. JAMA.1986; 256(20):2835
-2838.6. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome
among US adults: findings from the third National Health and Nutrition
Examination Survey. JAMA.2002; 287(3):356
-359.7. Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management
of the metabolic syndrome: an American Heart Association/National Heart, Lung,
and Blood Institute Scientific Statement. Circulation.2005; 112(17):2735
-2752.8. Sharrett AR, Ballantyne CM, Coady SA, et al. Coronary heart disease
prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a),
apolipoproteins A-I and B, and HDL density subfractions: the Atherosclerosis
Risk in Communities (ARIC) Study. Circulation.2001; 104(10):1108
-1113.9. Chapman MJ, Assmann G, Fruchart JC, Shepherd J, Sirtori C. Raising
high-density lipoprotein cholesterol with reduction of cardiovascular risk:
the role of nicotinic acid—a position paper developed by the European
Consensus Panel on HDL-C. Curr Med Res Opin.2004; 20(8):1253
-1268.10. Gordon DJ, Rifkind BM. High-density lipoprotein—the clinical
implications of recent studies. N Engl J Med.1989; 321(19):1311
-1316.11. Assmann G, Gotto AM Jr. HDL cholesterol and protective factors in
atherosclerosis. Circulation.2004; 109(23)(suppl 1):iii8
-III14.12. Lewis GF, Rader DJ. New insights into the regulation of HDL
metabolism and reverse cholesterol transport. Circ
Res.2005; 96(12):1221
-1232.13. Curtiss LK, Volenta DT, Hime NJ, Rye KA. What is so special about
apolipoprotein AI in reverse cholesterol transport? Arterioscler
Thromb Vasc Biol.2006; 26(1):12
-19.14. Meyers CD, Kashyap ML. Pharmacologic elevation of high-density
lipoproteins: recent insights on mechanism of action and atherosclerosis
protection. Curr Opin Cardiol.2004; 19(4):366
-373.15. Shah PK, Kaul S, Nilsson J, Cercek B. Exploiting the vascular
protective effects of high-density lipoprotein and its apolipoproteins: an
idea whose time for testing is coming, part I.
Circulation.2001; 104 (19):2376
-2383.16. Navab M, Ananthramaiah GM, Reddy ST, et al. The oxidation
hypothesis of atherogenesis: the role of oxidized phospholipids and HDL.
J Lipid Res.2004; 45(6):993
-1007.17. Linsel-Nitschke P, Tall AR. HDL as a target in the treatment of
atherosclerotic cardiovascular disease. Nat Rev Drug
Discov.2005; 4(3):193
-205.18. Ansell BJ, Watson KE, Fogelman AM, Navab M, Fonarow GC.
High-density lipoprotein function recent advances. J Am Coll
Cardiol.2005; 46(10):1792
-1798.19. Ansell BJ, Fonarow GC, Fogelman AM. High-density lipoprotein: is it
always atheroprotective? Curr Atheroscler Rep.2006; 8(5):405
-411.20. Zheng L, Nukuna B, Brennan ML, et al. Apolipoprotein A-I is a
selective target for myeloperoxidase-catalyzed oxidation and functional
impairment in subjects with cardiovascular disease. J Clin
Invest.2004; 114(4):529
-541.21. Ansell BJ, Navab M, Hama S, et al. Inflammatory/antiinflammatory
properties of high-density lipoprotein distinguish patients from control
subjects better than high-density lipoprotein cholesterol levels and are
favorably affected by simvastatin treatment.
Circulation.2003; 108(22):2751
-2756.22. Asztalos BF, Collins D, Cupples LA, et al. Value of high-density
lipoprotein (HDL) subpopulations in predicting recurrent cardiovascular events
in the Veterans Affairs HDL Intervention Trial. Arterioscler Thromb
Vasc Biol.2005; 25(10):2185
-2191.23. Rader DJ. Apolipoprotein A-I mimetic peptide D-4F in humans with
CHD improves HDL anti-inflammatory function after a single dose [abstract].
Circulation.2006; 114(suppl):II
-288.24. Kraus WE, Houmard JA, Duscha BD, et al. Effects of the amount and
intensity of exercise on plasma lipoproteins. N Engl J
Med.2002; 347(19):1483
-1492.25. Roberts CK, Ng C, Hama S, Eliseo AJ, Barnard RJ. Effect of a
short-term diet and exercise intervention on inflammatory/anti-inflammatory
properties of HDL in overweight/obese men with cardiovascular risk factors.
J Appl Physiol.2006; 101(6):1727
-1732.26. Khurana M, Sharma D, Khandelwal PD. Lipid profile in smokers and
tobacco chewers—a comparative study. J Assoc Physicians
India.2000; 48(9):895
-897.27. Maeda K, Noguchi Y, Fukui T. The effects of cessation from
cigarette smoking on the lipid and lipoprotein profiles: a meta-analysis.
Prev Med.2003; 37(4):283
-290.28. Weisweiler P. Plasma lipoproteins and lipase and
lecithin:cholesterol acyltransferase activities in obese subjects before and
after weight reduction. J Clin Endocrinol Metab.1987; 65(5):969
-973.29. Dattilo AM, Kris-Etherton PM. Effects of weight reduction on blood
lipids and lipoproteins: a meta-analysis. Am J Clin
Nutr.1992; 56(2):320
-328.30. Burke V, Beilin LJ, Cutt HE, Mansour J, Williams A, Mori TA. A
lifestyle program for treated hypertensives improved health-related behaviors
and cardiovascular risk factors, a randomized controlled trial. J
Clin Epidemiol.2007; 60(2):133
-141.31. Després JP, Golay A, Sjostrom L. Effects of rimonabant on
metabolic risk factors in overweight patients with dyslipidemia. N
Engl J Med.2005; 353(20):2121
-2134.32. Pi-Sunyer FX, Aronne LJ, Heshmati HM, Devin J, Rosenstock J. Effect
of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic
risk factors in overweight or obese patients: RIO-North America: a randomized
controlled trial. JAMA.2006; 295(7):761
-775.33. Valmadrid CT, Klein R, Moss SE, Klein BE, Cruick-shanks KJ. Alcohol
intake and the risk of coronary heart disease mortality in persons with
older-onset diabetes mellitus. JAMA.1999; 282(3):239
-246.34. Gaziano JM, Buring JE, Breslow JL, et al. Moderate alcohol intake,
increased levels of high-density lipoprotein and its subfractions, and
decreased risk of myocardial infarction. N Engl J Med.1993; 329(25):1829
-1834.35. Rimm EB, Moats C. Alcohol and coronary heart disease: drinking
patterns and mediators of effect. Ann Epidemiol.2007; 17(5)(suppl):S3
-S7.36. Nicholls SJ, Lundman P, Harmer JA, et al. Consumption of saturated
fat impairs the anti-inflammatory properties of high-density lipoproteins and
endothelial function. J Am Coll Cardiol.2006; 48(4):715
-720.37. Mensink RP, Zock PL, Kester AD, Katan MB. Effects of dietary fatty
acids and carbohydrates on the ratio of serum total to HDL cholesterol and on
serum lipids and apolipoproteins: a meta-analysis of 60 controlled trials.
Am J Clin Nutr.2003; 77(5):1146
-1155.38. Kromhout D, Bosschieter EB, de Lezenne Coulander C. The inverse
relation between fish consumption and 20-year mortality from coronary heart
disease. N Engl J Med.1985; 312(19):1205
-1209.39. Harris WS. n-3 fatty acids and serum lipoproteins: human studies.
Am J Clin Nutr.1997; 65(5) (suppl):1645S
-1654S.40. Slyper A, Jurva J, Pleuss J, Hoffmann R, Gutterman D. Influence of
glycemic load on HDL cholesterol in youth. Am J Clin
Nutr.2005; 81(2):376
-379.41. Ornish D, Scherwitz LW, Billings JH, et al. Intensive lifestyle
changes for reversal of coronary heart disease. JAMA.1998; 280(23):2001
-2007.42. National Cholesterol Education Program (NCEP) Expert Panel on
Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults
(Adult Treatment Panel III). Executive Summary of The Third Report of the
National Cholesterol Education Program (NCEP) Expert Panel on Detection,
Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment
Panel III). JAMA.2001; 285(19):2486
-2497.43. Kodama S, Tanaka S, Saito K, et al. Effect of aerobic exercise
training on serum levels of high-density lipoprotein cholesterol: a
meta-analysis. Arch Intern Med.2007; 167(10):999
-1008.44. Clofibrate and niacin in coronary heart disease.
JAMA.1975; 231(4):360
-381.45. Blankenhorn DH, Azen SP, Crawford DW, et al. Effects of
colestipol-niacin therapy on human femoral atherosclerosis.
Circulation.1991; 83(2):438
-447.46. Blankenhorn DH, Nessim SA, Johnson RL, Sanmarco ME, Azen SP,
Cashin-Hemphill L. Beneficial effects of combined colestipol-niacin therapy on
coronary atherosclerosis and coronary venous bypass grafts.
JAMA.1987; 257(23):3233
-3240.47. Blankenhorn DH, Selzer RH, Crawford DW, et al. Beneficial effects
of colestipol-niacin therapy on the common carotid artery: two- and four-year
reduction of intima-media thickness measured by ultrasound.
Circulation.1993; 88(1):20
-28.48. Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin,
antioxidant vitamins, or the combination for the prevention of coronary
disease. N Engl J Med.2001; 345(22):1583
-1592.49. Brown G, Albers JJ, Fisher LD, et al. Regression of coronary artery
disease as a result of intensive lipid-lowering therapy in men with high
levels of apolipoprotein B. N Engl J Med.1990; 323(19):1289
-1298.50. Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in
Coronary Drug Project patients: long-term benefit with niacin. J Am
Coll Cardiol.1986; 8(6):1245
-1255.51. Carlson LA, Rosenhamer G. Reduction of mortality in the Stockholm
Ischaemic Heart Disease Secondary Prevention Study by combined treatment with
clofibrate and nicotinic acid. Acta Med Scand.1988; 223(5):405
-418.52. Cashin-Hemphill L, Mack WJ, Pogoda JM, Sanmarco ME, Azen SP,
Blankenhorn DH. Beneficial effects of colestipol-niacin on coronary
atherosclerosis: a 4-year follow-up. JAMA.1990; 264(23):3013
-3017.53. Haskell WL, Alderman EL, Fair JM, et al. Effects of intensive
multiple risk factor reduction on coronary atherosclerosis and clinical
cardiac events in men and women with coronary artery disease: the Stanford
Coronary Risk Intervention Project (SCRIP).
Circulation.1994; 89(3):975
-990.54. Taylor AJ, Lee HJ, Sullenberger LE. The effect of 24 months of
combination statin and extended-release niacin on carotid intima-media
thickness: ARBITER 3. Curr Med Res Opin.2006; 22(11):2243
-2250.55. Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial
Biology for the Investigation of the Treatment Effects of Reducing Cholesterol
(ARBITER) 2: a double-blind, placebo-controlled study of extended-release
niacin on atherosclerosis progression in secondary prevention patients treated
with statins. Circulation.2004; 110(23):3512
-3517.56. Whitney EJ, Krasuski RA, Personius BE, et al. A randomized trial of
a strategy for increasing high-density lipoprotein cholesterol levels: effects
on progression of coronary heart disease and clinical events. Ann
Intern Med.2005; 142(2):95
-104.57. Frick MH, Elo O, Haapa K, et al. Helsinki Heart Study:
primary-prevention trial with gemfibrozil in middle-aged men with
dyslipidemia: safety of treatment, changes in risk factors, and incidence of
coronary heart disease. N Engl J Med.1987; 317(20):1237
-1245.58. Rubins HB, Robins SJ, Collins D, et al; Veterans Affairs
High-Density Lipoprotein Cholesterol Intervention Trial Study Group.
Gemfibrozil for the secondary prevention of coronary heart disease in men with
low levels of high-density lipoprotein cholesterol. N Engl J
Med.1999; 341(6):410
-418.59. Trial of clofibrate in the treatment of ischaemic heart disease:
five-year study by a group of physicians of the Newcastle upon Tyne region.
Br Med J.1971; 4(5790):767
-775.60. Ischaemic heart disease: a secondary prevention trial using
clofibrate: report by a research committee of the Scottish Society of
Physicians. Br Med J.1971; 4(5790):775
-784.61. A co-operative trial in the primary prevention of ischaemic heart
disease using clofibrate: report from the Committee of Principal
Investigators. Br Heart J.1978; 40(10):1069
-1118.62. WHO cooperative trial on primary prevention of ischaemic heart
disease with clofibrate to lower serum cholesterol: final mortality follow-up:
report of the Committee of Principal Investigators.
Lancet.1984; 2(8403):600
-604.63. Secondary prevention by raising HDL cholesterol and reducing
triglycerides in patients with coronary artery disease: the Bezafibrate
Infarction Prevention (BIP) study. Circulation.2000; 102(1):21
-27.64. Effect of fenofibrate on progression of coronary-artery disease in
type 2 diabetes: the Diabetes Atherosclerosis Intervention Study, a randomised
study. Lancet.2001; 357(9260):905
-910.65. Ericsson CG, Hamsten A, Nilsson J, Grip L, Svane B, de Faire U.
Angiographic assessment of effects of bezafibrate on progression of coronary
artery disease in young male postinfarction patients.
Lancet.1996; 347(9005):849
-853.66. Frick MH, Syvanne M, Nieminen MS, et al; Lopid Coronary Angiography
Trial (LOCAT) Study Group. Prevention of the angiographic progression of
coronary and vein-graft atherosclerosis by gemfibrozil after coronary bypass
surgery in men with low levels of HDL cholesterol.
Circulation.1997; 96(7):2137
-2143.67. Meade T, Zuhrie R, Cook C, Cooper J. Bezafibrate in men with lower
extremity arterial disease: randomised controlled trial.
BMJ.2002; 325(7373):1139
.68. Jones PH, Davidson MH, Stein EA, et al. Comparison of the efficacy
and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin
across doses (STELLAR* Trial). Am J
Cardiol.2003; 92(2):152
-160.69. Asztalos BF, Horvath KV, McNamara JR, Roheim PS, Rubinstein JJ,
Schaefer EJ. Comparing the effects of five different statins on the HDL
subpopulation profiles of coronary heart disease patients.
Atherosclerosis.2002; 164(2):361
-369.70. Deakin S, Leviev I, Guernier S, James RW. Simvastatin modulates
expression of the PON1 gene and increases serum paraoxonase: a role for sterol
regulatory element-binding protein-2. Arterioscler Thromb Vasc
Biol.2003; 23(11):2083
-2089.71. Klerkx AH, de Grooth GJ, Zwinderman AH, Jukema JW, Kuivenhoven JA,
Kastelein JJ. Cholesteryl ester transfer protein concentration is associated
with progression of atherosclerosis and response to pravastatin in men with
coronary artery disease (REGRESS). Eur J Clin Invest.2004; 34(1):21
-28.72. Martin G, Duez H, Blanquart C, et al. Statin-induced inhibition of
the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I.
J Clin Invest.2001; 107(11):1423
-1432.73. Nicholls SJ, Tuzcu EM, Sipahi I, et al. Statins, high-density
lipoprotein cholesterol, and regression of coronary atherosclerosis.
JAMA.2007; 297(5):499
-508.74. Heart Protection Study Collaborative Group. MRC/BHF Heart
Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk
individuals: a randomised placebo-controlled trial.
Lancet.2002; 360(9326):7
-22.75. Ansell BJ. Rationale for combination therapy with statin drugs in
the treatment of dyslipidemia. Curr Atheroscler Rep.2005; 7(1):29
-33.76. Bavirti S, Ghanaat F, Tayek JA. Peroxisome proliferator-activated
receptor-gamma agonist increases both low-density lipoprotein cholesterol
particle size and small high-density lipoprotein cholesterol in patients with
type 2 diabetes independent of diabetic control. Endocr
Pract.2003; 9(6):487
-493.77. Dormandy JA, Charbonnel B, Eckland DJ, et al. Secondary prevention
of macrovascular events in patients with type 2 diabetes in the PROactive
Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a
randomised controlled trial. Lancet.2005; 366(9493):1279
-1289.78. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of
myocardial infarction and death from cardiovascular causes. N Engl
J Med.2007; 356(24):2457
-2471.79. Kashyap ML, McGovern ME, Berra K, et al. Longterm safety and
efficacy of a once-daily niacin/lovastatin formulation for patients with
dyslipidemia. Am J Cardiol.2002; 89(6):672
-678.80. Keech A, Simes RJ, Barter P, et al. Effects of long-term
fenofibrate therapy on cardiovascular events in 9795 people with type 2
diabetes mellitus (the FIELD study): randomised controlled trial.
Lancet. 2005;366
(9500):1849
-1861.81. Nissen SE, Tardif JC, Nicholls SJ, et al. Effect of torcetrapib on
the progression of coronary atherosclerosis. N Engl J
Med.2007; 356(13):1304
-1316.82. Kastelein JJ, van Leuven SI, Burgess L, et al. Effect of
torcetrapib on carotid atherosclerosis in familial hypercholesterolemia.
N Engl J Med.2007; 356(16):1620
-1630.83. Nissen SE, Tsunoda T, Tuzcu EM, et al. Effect of recombinant ApoA-I
Milano on coronary atherosclerosis in patients with acute coronary syndromes:
a randomized controlled trial. JAMA.2003; 290(17):2292
-2300.84. Tardif JC, Gregoire J, L'Allier PL, et al. Effects of reconstituted
high-density lipoprotein infusions on coronary atherosclerosis: a randomized
controlled trial. JAMA.2007; 297(15):1675
-1682.85. Elam MB, Hunninghake DB, Davis KB, et al. Effect of niacin on lipid
and lipoprotein levels and glycemic control in patients with diabetes and
peripheral arterial disease: the ADMIT study: a randomized trial.
JAMA.2000; 284(10):1263
-1270.86. Grundy SM, Vega GL, McGovern ME, et al. Efficacy, safety, and
tolerability of once-daily niacin for the treatment of dyslipidemia associated
with type 2 diabetes: results of the assessment of diabetes control and
evaluation of the efficacy of Niaspan trial. Arch Intern
Med.2002; 162(14):1568
-1576.87. Forrester JS, Shah PK. Emerging strategies for increasing
high-density lipoprotein. Am J Cardiol.2006; 98(11):1542
-1549.88. Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E,
Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein
metabolism. Circulation.1998; 98(19):2088
-2093.89. Després JP, Lemieux I, Robins SJ. Role of fibric acid
derivatives in the management of risk factors for coronary heart disease.
Drugs.2004; 64(19):2177
-2198.90. Császár A. Hypertriglyceridemia, the coronary heart
disease risk marker "solved." Acta Physiol
Hung.2005; 92(2):109
-120.91. Action to Control Cardiovascular Risk in Diabetes (ACCORD)
[NCT00000620].
http://www.clinicaltrials.gov/ct/show/NCT00000620?order=1.
Accessibility verified July 19, 2007.92. Cheng K, Wu TJ, Wu KK, et al. Antagonism of the prostaglandin D2
receptor 1 suppresses nicotinic acid-induced vasodilation in mice and humans.
Proc Natl Acad Sci U S A.2006; 103(17):6682
-6687.93. Tall AR. Plasma cholesteryl ester transfer protein. J
Lipid Res.1993; 34(8):1255
-1274.94. Matsuura F, Wang N, Chen W, Jiang XC, Tall AR. HDL from
CETP-deficient subjects shows enhanced ability to promote cholesterol efflux
from macrophages in an apoE- and ABCG1-dependent pathway. J Clin
Invest.2006; 116(5):1435
-1442.95. Barter PJ, Brewer HB Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall
AR. Cholesteryl ester transfer protein: a novel target for raising HDL and
inhibiting atherosclerosis. Arterioscler Thromb Vasc
Biol.2003; 23(2):160
-167.96. Curb JD, Abbott RD, Rodriguez BL, et al. A prospective study of
HDL-C and cholesteryl ester transfer protein gene mutations and the risk of
coronary heart disease in the elderly. J Lipid Res.2004; 45(5):948
-953.97. Zhong S, Sharp DS, Grove JS, et al. Increased coronary heart
disease in Japanese-American men with mutation in the cholesteryl ester
transfer protein gene despite increased HDL levels. J Clin
Invest.1996; 97(12):2917
-2923.98. Kuivenhoven JA, de Grooth GJ, Kawamura H, et al. Effectiveness of
inhibition of cholesteryl ester transfer protein by JTT-705 in combination
with pravastatin in type II dyslipidemia. Am J
Cardiol.2005; 95(9):1085
-1088.99. Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ,
Fogelman AM. Mechanisms of disease: proatherogenic HDL—an evolving
field. Nat Clin Pract Endocrinol Metab.2006; 2(9):504
-511.100. Okamoto H, Yonemori F, Wakitani K, Minowa T, Maeda K, Shinkai H. A
cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in
rabbits. Nature.2000; 406(6792):203
-207.101. Huang Z, Inazu A, Nohara A, Higashikata T, Mabuchi H. Cholesteryl
ester transfer protein inhibitor (JTT-705) and the development of
atherosclerosis in rabbits with severe hypercholesterolaemia. Clin
Sci (Lond).2002; 103(6):587
-594.102. de Grooth GJ, Kuivenhoven JA, Stalenhoef AF, et al. Efficacy and
safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in
humans: a randomized phase II dose-response study.
Circulation.2002; 105(18):2159
-2165.103. Kuwabara K, Murakami K, Todo M, et al. A novel selective peroxisome
proliferator-activated receptor alpha agonist,
2-methyl-c-5-[4-[5-methyl-2-(4-methylphenyl)-4-oxazolyl]butyl]-1,3-dioxane-r-2-carboxylic
acid (NS-220), potently decreases plasma triglyceride and glucose levels and
modifies lipoprotein profiles in KK-Ay mice. J Pharmacol Exp
Ther.2004; 309(3):970
-977.104. Oliver WR Jr, Shenk JL, Snaith MR, et al. A selective peroxisome
proliferator-activated receptor delta agonist promotes reverse cholesterol
transport. Proc Natl Acad Sci U S A.2001; 98(9):5306
-5311.105. Navab M, Anantharamaiah GM, Hama S, et al. Oral administration of
an Apo A-I mimetic peptide synthesized from D-amino acids dramatically reduces
atherosclerosis in mice independent of plasma cholesterol.
Circulation.2002; 105(3):290
-292.106. Navab M, Anantharamaiah GM, Reddy ST, et al. Oral D-4F causes
formation of pre-beta high-density lipoprotein and improves high-density
lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from
macrophages in apolipoprotein E-null mice.
Circulation.2004; 109(25):3215
-3220.107. Navab M, Hama S, Hough G, Fogelman AM. Oral synthetic phospholipid
(DMPC) raises high-density lipoprotein cholesterol levels, improves
high-density lipoprotein function, and markedly reduces atherosclerosis in
apolipoprotein E-null mice. Circulation.2003; 108(14):1735
-1739.108. Burgess JW, Neville TA, Rouillard P, Harder Z, Beanlands DS, Sparks
DL. Phosphatidylinositol increases HDL-C levels in humans. J Lipid
Res.2005; 46(2):350
-355.109. Rodrigueza WV, Klimuk SK, Pritchard PH, Hope MJ. Cholesterol
mobilization and regression of atheroma in cholesterol-fed rabbits induced by
large unila-mellar vesicles. Biochim Biophys Acta.1998; 1368(2):306
-320.110. Treatment of HDL to Reduce the Incidence of Vascular Events
HPS2-Thrive [NCT00461630].
http://www.clinicaltrials.gov/ct/show/NCT00461630?order=1.
Accessibility verified July 18, 2007.111. Brousseau ME, Schaefer EJ, Wolfe ML, et al. Effects of an inhibitor
of cholesteryl ester transfer protein on HDL cholesterol. N Engl J
Med.2004; 350(15):1505
-1515.112. Clark RW, Sutfin TA, Ruggeri RB, et al. Raising high-density
lipoprotein in humans through inhibition of cholesteryl ester transfer
protein: an initial multidose study of torcetrapib. Arterioscler
Thromb Vasc Biol.2004; 24(3):490
-497.113. Morehouse LA, Sugarman ED, Bourassa PA, et al. Inhibition of CETP
activity by torcetrapib reduces susceptibility to diet-induced atherosclerosis
in New Zealand White rabbits. J Lipid Res.2007; 48(6):1263
-1272.114. Tall AR, Yvan-Charvet L, Wang N. The failure of torcetrapib: was it
the molecule or the mechanism? Arterioscler Thromb Vasc
Biol.2007; 27(2):257
-260.115. Navab M, Anantharamaiah GM, Reddy ST, et al. Apolipoprotein A-I
mimetic peptides. Arterioscler Thromb Vasc Biol.2005; 25(7):1325
-1331.116. Nissen SE, Nicholls SJ, Wolski K, et al. Effects of a potent and
selective PPAR-alpha agonist in patients with atherogenic dyslipidemia or
hypercholesterolemia: two randomized controlled trials.
JAMA.2007; 297(12):1362
-1373.117. GW501516 in Subjects Who Have Low Level of High-Density Lipoprotein
Cholesterol [NCT00158899].
http://www.clinicaltrials.gov/ct/show/NCT00158899?order=1.
Accessibility verified July 26, 2007.118. Buse JB, Rubin CJ, Frederich R, et al. Muraglitazar, a dual
(alpha/gamma) PPAR activator: a randomized, double-blind, placebo-controlled,
24-week monotherapy trial in adult patients with type 2 diabetes.
Clin Ther.2005; 27(8):1181
-1195.119. Skrumsager BK, Nielsen KK, Muller M, Pabst G, Drake PG, Edsberg B.
Ragaglitazar: the pharmacokinetics, pharmacodynamics, and tolerability of a
novel dual PPAR alpha and gamma agonist in healthy subjects and patients with
type 2 diabetes. J Clin Pharmacol.2003; 43(11):1244
-1256.120. Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and
major adverse cardiovascular events in patients with type 2 diabetes mellitus.
JAMA.2005; 294(20):2581
-2586.121. Haffner SM. Dyslipidemia management in adults with diabetes.
Diabetes Care. 2004;(27)
(suppl 1): S68-S71.122. Navab M, Ananthramaiah GM, Reddy ST, et al. The double jeopardy of
HDL. Ann Med.2005; 37(3):173
-178.123. Castro GR, Fielding CJ. Early incorporation of cell-derived
cholesterol into pre-beta-migrating high-density lipoprotein.
Biochemistry.1988; 27(1):25
-29.124. Nissen SE, Tuzcu EM, Brewer HB, et al. Effect of ACAT inhibition on
the progression of coronary atherosclerosis. N Engl J
Med.2006; 354(12):1253
-1263.
ARTICLE EN RAPPORT
Cette semaine dans le JAMA
JAMA. 2007;298:715.
Texte Complet
|