The Secrets of a Successful Clinical Trial: Compliance, Compliance, and Compliance
- 1 Semmelweis University, Department of Psychiatry and Psychotherapy, Budapest, Hungary
- 2 Nathan Kline Institute for Psychiatric Research, Orangeburg, New York 10962
- 3 Division of Pharmacotherapies and Medical Consequences of Drug Abuse, National Institute on Drug Abuse, National Institutes of Health, Bethesda, Maryland 20892
Technological advances have brought unprecedented rigor to target interrogation, providing the means for linking a specific target to a disease, for establishing that a medication occupies and engages its target at appropriate doses, and for marshaling pharmacokinetic information in order to predict optimum dosing prior to initiating proof-of-efficacy studies. Nevertheless, drug development remains a high cost and risky undertaking (1, 2), and if a compound does not elicit the desired efficacy signal in the clinic, blame is most often cast on the inability to appropriately interrogate the targets, and particularly on the lack of translational fidelity of animal models (3). Critical “go/no-go” decisions, such as first-in-man studies to obtain information about the pharmacokinetics, safety, and tolerability of a drug, as well as imaging studies to measure target engagement, are typically executed in well controlled clinical settings, with subjects dosed in clinic. In contrast, once a compound has progressed to efficacy trials, both economic and logistic considerations most often dictate that studies will be done in an outpatient population. The current nature of commercially sponsored clinical trials, including strict sponsor-imposed timelines and competition among sites for eligible subjects, has created an environment that can provide significant economic rewards for both the subject and trial site (4). An unfortunate byproduct of this environment is the “professional subject” (4). Although more commonly considered to be a phase 1 phenomenon (4), efficacy trials also attract professional subjects, particularly when entry criteria and endpoints are “soft,” such as trials using subjective rating scales which can be “gamed.” It has become apparent that many subjects enrolling in clinical trials are simply not medication-compliant, with a significant proportion failing to take even a single dose of medication over the course of the study. In the following, we illustrate the effect of noncompliance on clinical trial outcome, and propose solutions, however imperfect, that can be readily implemented for more accurate assessment of drug safety and efficacy.
Our first illustration of the problem relates to a phase 1b safety and tolerability study in which patient noncompliance had the effect of masking a potential efficacy signal. The eight-week study in this instance, using the triple uptake inhibitor DOV 21947 (5), was conducted as a single-site outpatient trial. Subjects were randomized at approximately 2:1 (drug: placebo), and per protocol, blood was drawn at weeks 2, 4, 6, and 8. Any detection of DOV 21947 (or its lactam metabolite) was scored as evidence of medication compliance for the given visit, and three compliant visits provided the threshold for designating the subject as medication-compliant within the trial. Among the subjects randomized to the DOV 21947 arm, twenty-three of thirty-one subjects completed the study, with 70% of the subjects (16/23) meeting the criterion for medication compliance (Table 1). In marked contrast, compliance as measured by counting pill usage was greater than 92%. Admittedly, this comparison, along with the analysis we will give below, offers only a “snapshot” of compliance; it cannot be asserted that subjects without a single compliant visit never took the drug, nor can it determined that subjects with ”full compliance” [who, in this case, comprised 39% of subjects (9/23) at trial completion] took every dose of drug. However, given the blood levels predicted from phase 1a studies, where drug was administered in clinic (6) with an assay sensitivity of about 3 ng/ml, it is likely the consistent absence of drug or metabolite over 4 visits, indicates these individuals missed multiple doses.
aMedication Compliance in a Phase 1b Safety and Tolerability Study
A significant weight difference was noted between the medication-compliant cohort and the placebo group (Figure 1). It should be noted that this difference would have been masked if the effect of medication compliance had been ignored: a comparison between the placebo group and patients in the drug arm (i.e., without consideration of compliance) shows no difference regarding weight change. Although the weight effect was not particularly robust (a loss of approximately 1 kg in the compliant group, and a 2 kg difference from both the placebo and noncompliant groups, it should be noted that the data were generated in the absence of procedures (e.g., exercise and dietary guidelines) routinely used to assess weight loss as a primary outcome measure in clinical trials (7). Moreover, the weight loss in medication-compliant subjects fully dissipated within one week following the cessation of medication (Figure 1), indicating this effect was medication related. Rodent models of obesity have established that DOV 21947 elicits reductions in both food intake and animal weight (8). The effective dose range in these obesity models overlaps doses active in rodent models of behavioral despair (9), such as the forced swim and tail suspension tests, which are frequently employed to predict an antidepressant action (10). The weight loss signal observed in medication-compliant subjects proved valuable (6) for setting the doses of DOV 21947 in phase 2, where a robust antidepressant effect was subsequently evinced (11).
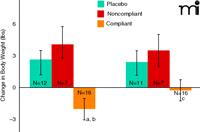
Change in body weight at study end point (left panel) and at one-week post-treatment (right panel) are shown. (ANCOVA among the three study groups; body weight at baseline and gender were applied as covariates. ap<0.0055 vs placebo; bp<0.0019 vs noncompliant; cp<0.044 vs noncompliant).
The second example that we offer to illustrate the problem of noncompliance in clinical studies comes from a phase 3 study of the analgesic, bicifadine. This norepinephrine- and serotonin-reuptake inhibitor had been demonstrated to produce analgesic actions in both preclinical (12) and clinical (13, 14) studies, where it was reported to be as efficacious as codeine in a model of dental pain and as effective as tramadol in reducing post-bunionectomy pain. In both the dental pain and bunionectomy studies, medications were administered in clinic. Based on these positive findings, a 634-patient, twelve-week trial was conducted, where three doses of drug were compared to placebo in moderate to severe chronic low back pain. In a planned exposure-response comparison in one study arm receiving 200 mg of drug twice daily, blood samples were obtained at study weeks 2, 4, 8, and 12. Compliance, as defined by the detection (lower limit of detection, > 0.5 ng/ml) of plasma bicifadine, never exceeded 67.5% of medicated patients, and at week 12, only slightly more than half of the subjects (53.6%) were judged compliant. No significant differences were observed among the four study arms in either the primary outcome measure [change in a visual analog scale (VAS) score for pain] or secondary outcome measures [including the Roland-Morris Disability Questionnaire (15); see below] at the end of the twelve weeks. Based on single- and multiple-dose phase 1 studies, the controlled release formulation of bicifadine used throughout the trials discussed here exhibited well-behaved pharmacokinetic properties. The Cmax and AUC values were linear with respect to dose and consistent among studies.
When medication compliance was considered in this phase 3 study, a wholly different picture of efficacy emerged. Those patients who manifested plasma bicifadine at study end (week 12) had a significantly greater reduction in pain scale scores (VAS) than placebo patients (Figure 2). Further stratification of “compliant” bicifadine-treated patients (into individuals with drug levels above or below 500 ng/ml) revealed that the greatest reduction in VAS among all study groups occurred in individuals with the higher level of bicifadine, and the change in VAS for the placebo group was identical to that for the noncompliant group. Only those placebo-treated patients who spent at least two weeks in the trial were selected for this analysis, thereby assuring a fair comparison with compliant subjects who, by definition, had to spend at least two weeks in the study. In addition, given the Cmax value (approximately 675 ng/ml) obtained following a single 200-mg dose, and a t1/2 of about four hours (14), a conservative prediction is that three consecutive doses would have to be missed (corresponding to the passage of approximately ten half-lives) before plasma levels of bicifadine could drop to undetectable levels.
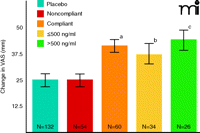
These data represent patients who received either a controlled release form of bicifadine (200 mg, twice daily) or placebo. The visual analog scale (VAS) ranges from 0 100 mm. For patient recruitment in the study reported here, baseline pain severity had to be at least 40 mm. Changes in VAS score was the primary endpoint measure; analysis of plasma bicifadine in this trial arm was a planned pharmacokinetic/pharmacodynamic comparison. VAS change scores as endpoint measures were compared with ANCOVA. Severity at baseline, gender and age were applied as covariates. The pattern of results shown here was largely replicated (data not shown) using the Roland-Morris Disability Questionnaire (15); see text for details. (ap<0.01 vs placebo or noncompliant groups; bp<0.05 vs placebo or noncompliant groups; cp<0.002 vs placebo or noncompliant groups).
A qualitatively similar pattern of drug efficacy emerged from a secondary endpoint in the study [i.e., the Roland-Morris Disability (RDQ) questionnaire, a widely used instrument to assess low back pain (15)]. Those patients who manifested detectable plasma levels of drug at study end were significantly improved compared to placebo-treated individuals. A stratification of RDQ responses was again observed in relation to plasma drug levels, with plasma levels above 500 ng/ml linked to a greater improvement as compared to the group with lower levels, the noncompliant group (i.e., individuals with no detectable levels of bicifadine), or the placebo group. As in the previous example with DOV 21947, compliance as determined through pill counts was ≥94% in each of the four arms (three drug doses and placebo) of this study. Also as in the DOV 21947 study, blood samples were taken intermittently, and can only be described as representing a “snapshot” of compliance. Thus, in the most extreme case, if a patient were to take only a single dose of drug immediately prior to each clinic visit, the subject could be classified as “compliant” for the trial. Nonetheless, even using this admittedly imperfect definition of “compliance,” a signal was detected in both the safety (Figure 1) and efficacy studies (Figure 2) described here.
Despite the insights obtained by analyzing patient “compliance,” some criticism against the incorporation of compliance analysis in evaluating drug safety and efficacy may well be raised. In particular, it could be argued that subjects become noncompliant for “real-world” reasons, such as either intolerable side effects or lack of efficacy, and that by relegating such study patients into a “noncompliant” category, we might inaccurately magnify predictions of drug safety and efficacy in the real-world population. However, in the phase 3 study described here (with a sufficient number of subjects in each arm to make the argument credible), it must be noted that the primary outcome measure was identical for both the placebo and noncompliant groups. In contrast, individuals who were compliant to some extent (i.e., manifesting detectable drug levels, however minimal, at study completion) separated from placebo on the primary outcome measure, and those in the cohort who were most compliant also manifested the largest change in primary endpoint. The assumption that lack of efficacy leads to noncompliance does not obtain in a phase 1 safety and tolerability study conducted in healthy volunteers; rejecting the argument that compliance should be monitored at every stage of clinical development can lead to missing potential efficacy signals (Figure 1). Further, confirmation that the drug discussed above proved safe and well tolerated in a larger cohort of depressed individuals (11) suggests that the noncompliance observed in a safety and tolerability study is not necessarily a reflection of side effects. Recognition of the “professional trial subject,” for example (see above), demands that we be aware of the social and economic realities of the current clinical trials environment.
Again, we do not mean to claim that the analyses presented here are definitive. The lack of a compliance measure in the placebo groups, for example, points out that our concerns require more rigorous investigation. Clearly, the a priori inclusion of a compliance measurement in all subjects (16) provides a compelling demonstration that monitoring medication compliance can be used to evince an efficacy signal. Thus, snapshots of compliance, achievable through various means of patient monitoring, can be brought to double-blind, placebo-controlled studies (16). The failure to acknowledge the effects of noncompliance on clinical data, moreover, could result in a “no go” decision for a compound, and sound the death knell for the small company capitalized for “one shot on goal.” Noncompliance is also relevant in assessing adverse events in clinical trials, before the development cycle of a medication might culminate in potentially disastrous economic and health consequences. In the real world, the rate of noncompliance to prescribed medication has been estimated as high as 50%, resulting in 89,000 premature deaths and a cost exceeding $100 billion annually (17). Admittedly, our discussion of noncompliance in clinical trials is not intended to address the problem with respect to prescribed medication at large.
In a meta-analysis of 192 publications summarizing randomized controlled trials in six chronic conditions, Gossec and colleagues reported a median compliance of 93%, predominantly determined by pill count. This rate of compliance is strikingly similar to the values obtained by pill count in the eight-week, single-site safety study (≥92%) and the twelve-week, multisite efficacy study (≥94%) exemplified above. The significant disparities between compliance determined from pill count and the measurement of drug in biological fluids are striking, and indicate that the former measure is of limited value in assessing medication compliance.
Currently, most clinical trial data are analyzed using an intent-to-treat (ITT) population, recording data from all subjects, generally following either randomization or administration of the first dose of drug. Proponents of this “all-comers” approach argue that it better replicates the “real-world” situation, where factors such as compliance are not considered when addressing treatment efficacy. However, imposing selected aspects of the real-world situation onto the artificial environment of a double-blind, placebo-controlled trial can result in the flawed testing of a hypothesis. Proponents also argue that the ITT approach provides randomization that prevents biased allocation of subjects to treatment groups and accordingly prevents biased results. Nonetheless, the “all-comers” approach ignores the impact of medication compliance on outcome, giving subjects who dutifully take their medications no more weight than to subjects who fail to adhere to trial protocols (e.g., the “professional” trial subject). Because it appears to have a major impact on trial outcome, meaningful statistical analyses should account for medication compliance (at minimum, at the level of “post-adjustment” procedures) in order to better assess the potential safety and efficacy of experimental drugs.
At present, direct and real-time observation is the only reliable, albeit impractical, means of verifying medication compliance. An imperfect but highly practical alternative is measuring the presence of drug at regular intervals, with a pre-determined frequency of values used to stratify medication compliance. As an example, in a study requiring eight visits to the clinic, the presence of drug on seven of these visits, as measured in urine or blood, could be established as criterion for identifying a subject as medication-compliant; exact criteria would be dependent on the particular pharmacokinetic data and assay sensitivity. Admittedly, monitoring medication compliance in any rigorous fashion will add significantly to clinical trial costs. However, investments into compliance analysis could pay off by the ability to make better informed go/no go decisions, thus preventing later expenditures into a development plan that may over time prove to be untenable (19).
Acknowledgments
The authors were employed by DOV Pharmaceutical, Inc. , during the conduct of these studies. The authors have no financial interests in the compounds described here.
Footnotes
-
Author contributions
Wrote or contributed to the writing of the manuscript: Czobor and Skolnick.
- Copyright © 2011
References
Pál Czobor, PhD, is an Associate Professor at Semmelweis University Department of Psychiatry and Psychotherapy, Budapest. He also serves as a research scientist at the Nathan Kline Institute for Psychiatric Research, Orangeburg, New York. His principal areas of interest include biostatistics, clinical trial design, psychopharmacology, and electrophysiology. Email czo-bor{at}psych.sote.hu.

Phil Skolnick, PhD, DSc (hon), is the Director, Division of Pharmacotherapies and Medical Consequences of Drug Abuse, at the National Institute on Drug Abuse, National Institutes of Health. He is focused on the identification and development of medications to treat substance use disorders. Address correspondence to PS. Email Phil.Skolnick{at}nih.gov.


