Biochemical Mechanisms of Cyclosporine Neurotoxicity
- 1Department of Anesthesiology and 2Department of Pharmacology, University of Colorado Health Sciences Center, Denver, CO 80262; 3Department of Biopharmaceutical Sciences, University of California, San Francisco, CA 94143
- Address correspondence to NS. E-mail Natalie.Serkova{at}uchsc.edu; fax 303-315-1858.
Abstract
Proper management of chemotoxicity in transplant patients requires detailed knowledge of the biochemical mechanisms underlying immunosuppressant toxicity. Neurotoxicity is one of the most significant clinical side effects of the immunosuppressive undecapeptide cyclosporine, occurring at some degree in up to 60% of transplant patients. The clinical symptoms of cyclosporine-mediated neurotoxicity consist of decreased responsiveness, hallucinations, delusions, seizures, cortical blindness, and stroke-like episodes that mimic those clinical symptoms of mitochondrial encephalopathy. Clinical computed tomography (CT) and magnetic resonance imaging (MRI) studies have revealed a correlation between clinical symptoms of cyclosporine-mediated neurotoxicity and morphological changes in the brain, such as hypodensity of white matter, cerebral edema, metabolic encephalopathy, and hypoxic damages. Paradoxically, in animal models cyclosporine protects the brain from ischemia–reperfusion (I/R) injury. Interestingly, cyclosporine appears to mediate both neurotoxicity (under normoxic conditions) and I/R protection across the same range of drug concentration. Both toxicity and protection might arise from the intersection of cyclosporine with mitochondrial energy metabolism. This review addresses basic biochemical mechanisms of: 1) cyclosporine toxicity in normoxic brain, and 2) its protective effects in the same organ during I/R. The marked and unparallel potential of magnetic resonance spectroscopy (MRS) as a novel quantitative approach to evaluate metabolic drug toxicity is described.

Introduction
Since the discovery of cortisol in 1949, many immunosuppressive drugs have been introduced into clinical transplantation. Their mechanisms of action differ from each other and can be summarized as follows: 1) regulators of gene expression (e.g., glucocorticoids); 2) alkylating agents (e.g., cyclophosphamide); 3) inhibitors of de novo purine synthesis (e.g., azathioprine and mycophenolate mofetil); 4) inhibitors of de novo pyrimidine synthesis (e.g., leflunomide); 5) antibodies (e.g., basiliximab and antithymocyte globulin); and 6) inhibitors of kinases and phosphatases (e.g., cyclosporine, tacrolimus, sirolimus, and everolimus) (1, 2). Currently, the calcineurin inhibitor cyclosporine A (also termed CsA) functions as the cornerstone of many immunosuppressive protocols to prevent graft rejection following organ transplantation. Cyclosporine is a lipophilic cyclic undecapeptide derived from the fungus Tolypocladium inflatum gams and in the cell it binds to its cytosolic receptor, an immunophilin termed cyclophilin D. The complex cyclosporine–cyclophilin D then binds to the calmodulin-stimulated protein phosphatase calcineurin, inhibiting calcineurin’s serine–threonine phosphatase activity (3). By preventing calcineurin-mediated dephosphorylation, cyclosporine inhibits the translocation of the NFAT (nuclear factor of activated T cells) family of transcription factors from the cytoplasm to the nucleus of activated T cells. The NFAT group is involved in the transcriptional activation of the genes encoding interleukin (IL)-2, IL-4 and CD40L; thus, inhibition of the NFAT by cyclosporine results in a specific inhibition of interleukin production in the T cell.
The adverse effects of cyclosporine include acute and chronic nephrotoxicity, neurotoxicity, hypertension, and new-onset diabetes (4, 5). Second to renal toxicity, neurotoxicity is the most serious side effect with cyclosporine, affecting 25% to 59% of transplant patients. Since 1979, when the first report describing cyclosporine-associated tremor in transplant patients was published (6), a wide range of cyclosporine-related neurological side effects have been associated with all types of organ transplantation, as well as with autoimmune disorders (7–9). Neurotoxicity is more frequently associated with measured steady-state minimum blood concentrations (Cmin) that exceed recommended levels, but also occurs during long-term treatment when concentrations of cyclosporine in the blood are within the therapeutic target range. Surprisingly, cyclosporine ameliorates brain injury induced by cerebral ischemia–reperfusion (I/R) in vivo (10). The proposed mechanism of neuroprotection of cyclosporine relates the drug’s ability to regulate the mitochondrial permeability transition (MPT) pore (11).
Management strategies for toxicity in transplant patients require a detailed knowledge of the mechanisms underlying cyclosporine neurotoxicity; however, in contrast to the well-established immunosuppressive mechanisms of cyclosporine action, biochemical aspects of cyclosporine effects in the brain remain poorly understood. Many pathways for toxicity have been discussed without distinguishing the principal mechanisms. In general, one of the major reasons for previous problems in toxicodynamics was the lack of an appropriate technique that would allow simultaneous evaluation of biochemical processes within the cell. During the last three years, magnetic resonance spectroscopy (MRS) has emerged as a powerful tool to evaluate biochemical changes that result from drug toxicity. One of the advantages of MRS is its non-selectiveness; that is, a wide range of biochemical pathways can be screened simultaneously. Based on our previous work using a quantitative MRS approach (12–17), we show that both cyclosporine-induced neurotoxic metabolic toxicity as well as cyclosporine’s neuroprotective effects against I/R arise from the drug’s interactions with brain mitochondria and are directly related to the decrease of mitochondrial energy production via inhibition of the Krebs cycle and the subsequent activation of anaerobic glycolysis.
Early Steps in Transplantation and Immunosuppression
In the mid-twentieth century, the first successful organ transplants were undertaken; Joseph Murray performed the first successful kidney transplant in 1954, and received a Nobel Prize for his work, in 1990. Thomas Starzl performed the first successful liver transplant in 1967. The first synthetic immunosuppressive treatment was introduced in 1962 when azathioprine increased the probability for successful human transplants, again garnering a Nobel Prize for innovative pharmacological progress. During the last twenty years, important medical breakthroughs such as tissue typing and introduction of calcineurin inhibitor–based immunosuppression dramatically improved the survival rate for transplant recipients. In 1983, the discovery of cyclosporine ushered in the era of calcineurin-inhibitor therapy, and transplantation of extrarenal organs became routine, with excellent outcomes for livers, pancreas, hearts, and even lungs. To date, the major limiting factors and concerns in the area of human transplantation remain the shortage of donor organs and the long-term survival of grafts, partly compromised by cyclosporine toxicity.
Because acute rejection rates are satisfactorily low, the focus of interest in transplantation has shifted recently towards tolerability and long-term graft and patient survival. Although recent analyses indicate that the half-life of allografts has increased, long-term results are still not acceptable partly because of cyclosporine-related side effects. The most limiting side effect of calcineurin inhibitors is nephrotoxicity (4, 5), followed by neurotoxicity (7–9), hypertension, diabetes, and hyperlipidemia (4). Even though nephrotoxicity remains the biggest problem with cyclosporine treatment, cyclosporine-dependent neurotoxicity occurs in up to 60% of organ transplant patients and can lead to serious life-threatening conditions and to withdrawal of cyclosporine from the patient’s regimen. These adverse effects have led to several clinical studies designed to reduce the cyclosporine dosage, to discontinue cyclosporine, or even to start “naïve” transplant patients on cyclosporine-free immunosuppressive protocols (2, 18, 19); however, success has been limited. Studies performed in the absence of cyclosporine to evaluate other rejection-inhibiting protocols [i.e., sirolimus+azathioprine+prednisolone (18), or sirolimus+mycophenolate+mofetil+prednisolone (19)] during the early post-transplant period reported that 40% of patients required treatment for acute rejection. Therefore, cyclosporine use will remain essential in clinical transplantation medicine, and a basic understanding of its toxic mechanisms is required to reduce the incidence of cyclosporine-related adverse effects.
Clinical Neurotoxicity
Observations of acute neurotoxicity in conjunction with high concentrations of cyclosporine in blood were reported soon after cyclosporine’s introduction into clinical practice. In 1979, the first report of cyclosporine-induced tremor in kidney, pancreas, or liver transplant patients exhibiting increased cyclosporine blood concentration was published (6). Subsequently, severe neurotoxicity resulting from cyclosporine treatment has been frequently reported in kidney (20–25), liver (26–34), heart (35–40), lung (41–44), and bone-marrow recipients (45–53) as well as in cyclosporine-treated patients with autoimmune diseases (54, 55). Although most published cases of cyclosporine-mediated neurotoxicity are related to liver and bone marrow transplantations, the overall incidence of CNS complications are comparable among the different organ transplant groups. In a report comparing the incidence of cyclosporine CNS toxicity in 500 patients from various organ transplant groups, cerebrovascular complications were most frequent and were observed in 51% of the liver, 59% of the heart, 58% of the lung, 50% of the heart-lung, 49% of the kidney, and 44% of the bone marrow allografts, with no significant difference between the organ groups (56). Overall, cyclosporine-mediated neurotoxicity was more severe in pediatric patients than in adults (24, 25, 33, 47, 50, 51, 57). Severe CNS symptoms included cortical blindness, decreased responsiveness, hallucinations, delusions, seizures, aphasia, ataxia, and stroke-like episodes (Table 1⇓) (7–9). Minor symptoms include tremor, agitation, insomnia, anxiety, amnesia, headache, and paraesthesia (7–9). In almost all reported cases, no CNS abnormalities were detected prior to transplantation (i.e., cyclosporine treatment) (9, 58). Clinical computerized tomography (CT) and magnetic resonance imaging (MRI) studies demonstrated a correlation between clinical symptoms of cyclosporine-dependent neurotoxicity and morphological changes in the brain, such as hypodensity of white matter, cerebral edema, metabolic encephalopathy, and hypoxic damage (Table 1⇓) (20, 23, 26, 27, 30, 32, 36, 40, 45, 46, 49–52, 57–66). Additional careful examinations of weighted MRI scans revealed that cyclosporine causes an acute ischemic insult secondary to vascular spasm with resultant axonal swelling (48, 65, 67, 68). In general, MRI was more sensitive in discovering cyclosporine-related abnormalities in the brain of patients with clinical manifestations of cyclosporine neurotoxicity as compared to CT (28, 40, 43, 69). In addition, electroencephalography (EEG) on cyclosporine treated patients with neurological abnormalities showed diffuse and focal slowing, abnormal waveforms, and epileptiform discharges (40, 50, 57, 70).
Cyclosporine-Induced Neurological Complications
In earlier reports, the incidence of neurotoxicity was positively correlated with high cyclosporine trough levels (Cmin) (6, 20, 22, 26, 27, 50, 59, 71). Berden et al. (20) reported a patient who, after renal transplantation, went into a coma for forty-four days. Blood cyclosporine concentrations in this patient ranged from 600 to 1000 ng/ml (normal range is <200 ng/ml) (20). Hypodensity of white matter with axonal degeneration and demyelination on his CT images suggested leucoencephalopathy, which disappeared after cyclosporine withdrawal and clinical recovery. Another report described the case of a liver transplant recipient with even higher concentrations of cyclosporine in the blood (>2000 ng/ml) (59). The patient had major seizures and hallucinations under cyclosporine treatment. Hypodensity of white matter was detected with a massive hemorrhage in the cerebellum. The patient did not recover after cyclosporine withdrawal. In most cases, however, neurotoxic symptoms were reversed with a reduction of cyclosporine dose or withdrawal (7, 8, 27, 38). More recently, chronic neurotoxicity during long-term treatment has been reported when cyclosporine blood concentrations were in the therapeutic target range (7, 23, 28, 32, 36, 40, 43, 57, 61, 67, 69). In contrast to early “acute” cyclosporine neurotoxicity (marked by high cyclosporine trough levels), late “chronic” neurotoxicity (marked by normal cyclosporine blood concentrations) tends to be longer lasting and to persist even after discontinuation of cyclosporine therapy (29, 36, 62, 63, 67). In the late 1980s, after characterization of cyclosporine metabolism and the development of reliable and specific analytical methods to measure cyclosporine and its metabolites in blood, various publications reported high trough levels of cyclosporine metabolites in patients with CNS dysfunctions (21, 38, 70). Cyclosporine is metabolized by cytochrome P4503A in small intestine and liver to several mostly hydroxylated, demethylated, or cyclized metabolites. The metabolites reach higher concentrations in blood than does cyclosporine, and it was hypothesized that not cyclosporine but high metabolite blood-trough concentrations were responsible for CNS toxicity (21, 38, 70); however, the metabolite-induced neurotoxicity theory has not been confirmed by later studies.
The clinical predisposition and mechanisms of cyclosporine-induced neurotoxicity remain controversial and poorly understood. Hypomagnesemia, hypocholesteremia, the vasoactive agent endothelin, as well as hypertension may facilitate cyclosporine-induced CNS abnormalities (Table 1⇑). Early diagnosis of cyclosporine neurotoxicity is important, because it is reversible in its early stages but, if diagnosed too late, it becomes potentially irreversible and fatal.
Hypomagnesemia
Hypomagnesemia was one of the first metabolic abnormalities observed in cyclosporine-treated recipients with neurotoxic symptoms (53). Thirty-five marrow-graft recipients that developed neurological symptoms under cyclosporine treatment also exhibited leukoencephalopathy, seizures, cerebellar ataxia, tremor, and/or depression (53). This study established an inverse correlation between neurological symptoms and low concentrations of magnesium in plasma. Hypomagnesemia may be caused by cyclosporine-induced intracellular migration of magnesium and renal wasting (i.e., urinary removal of magnesium). However, several other authors have shown that hypomagnesemia is not a prerequisite for cyclosporine-induced neurological complications (7, 28, 30).
Hypocholesterolemia
It might be reasonable to suspect that hypocholesterolemia may be a predisposition factor for development of cyclosporine neurotoxicity early after transplantation. Cyclosporine is a highly lipophilic drug. In plasma, 40–60% is bound to cholesterol-containing lipoproteins, especially low-density lipoprotein (LDL). Low cholesterol concentrations lead to increased free concentrations of cyclosporine. Low cholesterol levels also lead to an increase in the amount of LDL receptors expressed on the cell membranes (7). In the brain, LDL receptors are expressed in astrocytes (at the blood–brain barrier); therefore, increased uptake of cyclosporine can lead to increased permeability of the blood–brain barrier as well as to damage of the white matter observed in cyclosporine-treated patients with CNS symptoms. However, in the long term, the contribution of hypocholesterolemia is probably limited because cyclosporine increases free plasma cholesterol concentrations (72, 73), and thus reduces the prevalence of hypocholesterolemia.
Vasoconstriction
One of the common side effects of cyclosporine treatment is vasoconstriction. Severe episodes, caused by a disturbance in endothelial control of the vascular tone, lead to ischemia. Endothelin-1 and decrease of nitric oxide concentrations play a key role in cyclosporine-induced vasoconstriction (7, 41, 49).
Hypertension
Cyclosporine inhibits nitric oxide production through a calcineurin-sensitive pathway regulating eNOS dephosphorylation, which leads to hypertension (74). NO contributes to vasorelaxation, rather than to vasoconstriction (i.e., hypertension). Thus, Schwartz et al. (63) have proposed that hypertension is the major cause of cyclosporine-induced neurotoxicity; however, in many clinical scenarios, patients exhibiting clinical signs of neurotoxicity had normal arterial blood pressure, or neurotoxicity was developed prior to the clinical onset of hypertension (9).
Cyclosporine Neurotoxicity and Mitochondrial Encephalopathies
Interestingly, the clinical symptoms of cyclosporine neurotoxicity mimic those of mitochondrial encephalopathy (75, 76) (Table 2⇓). Mitochondrial encephalopathies (ME) are a heterogeneous group of metabolic diseases characterized by mitochondrial malfunction that leads to cellular energy failure with increased lactate production. Out of the morass of clinical reports, two ME syndromes seem distinctive (76, 77): 1) myoclonus epilepsy with tagged-red fibers (MERRF) (77), and 2) mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Over the last decade, the underlying genetic bases of MELAS and MERRF and two neurodegenerative diseases (i.e., Huntington Disease and Friedrich ataxia) have been identified (75–77). Despite these diseases’ different clinical features, the fact that neurons are highly dependent on oxidative energy metabolism suggests a similar pathogenetic mechanism underlying these disorders: dysfunctions in mitochondrial energy metabolism. Classifications of ME based on clinical or histopathological characteristics are unsatisfactory; therefore, MEs are now best classified according to the underlying biochemical defect. Biochemical manifestations of ME include a decrease in activity of NADH–coenzyme Q reductase (complex I), succinate–CoQ reductase (complex II), succinate–cytochrome c reductase (complex II–III), and cytochrome c oxidase (complex IV). The most prominent biochemical findings also include pyruvate dehydrogenase (PDH) deficiency, low blood pH, and increased blood lactate. Decreased activity of succinate dehydrogenase (SDH) and the subsequent increase in brain concentrations of succinate evaluated by in vivo 1H-MRS are less common metabolic findings, usually related to leukoencephalopathies (78). Impaired energy metabolism (decreased phosphocreatine to inorganic phosphate ratio) as well as increased lactate concentrations were found in the cerebral cortex of Huntington Disease patients by in vivo 31P- and 1H-MRS, respectively (79).
Comparing Symptoms of Cyclosporine-Induced Neurotoxicity and Mitochondrial Encephalomyopathies
The most compelling data for a close correspondence between cyclosporine-induced neurotoxicity and mitochondrial encephalopathies were reported in lung transplant recipients under cyclosporine treatment who developed metabolic myopathies as a cause of exercise limitation (44). These patients had impaired maximal exercise capacity because of a disorder of peripheral oxygen utilization. Additionally, epileptic seizures are often the first clinical symptom of many mitochondrial disorders involving the CNS and fully mimic those observed in cyclosporine-treated patients. Partial seizures are frequent in MELAS; generalized seizures are a cardinal symptom of the MERRF syndrome.
Metabolic Neurotoxicity of Cyclosporine
Application of MRS and MRS Based Metabonomics
Currently, many mechanisms have been proposed for cyclosporine-related neurotoxicity; however, cause-effect relationships are mostly unclear and the basic biochemical mechanism underlying cyclosporine neurotoxicity is very controversial. The living cell, as well as the entire organism, can be viewed through several levels of organization (Table 3⇓), which include: 1) genetics and gene expression (i.e., genomics and transcriptomics), 2) protein and signaling (i.e., proteomics), 3) metabolic control and regulation at the single-cell level (i.e., metabolomics), and 4) in the intact system at multiple levels over time (i.e., metabonomics). All pathophysiological stimuli and perturbations result in disturbances in concentrations, binding or fluxes of endogenous metabolites, either by direct chemical reaction or by changes in activation or expression of key enzymes or nucleic acids that control metabolism. Therefore, the assessment of metabolism in organs (either in the tissue itself or in body fluids surrounding it) can provide reliable information about toxic manifestations following pathophysiological stimuli (e.g., drug toxicity) (80, 81). The most likely reason for our incomplete understanding of the biochemical mechanisms involved in cyclosporine neurotoxicity is that conventional biochemical, enzymatic, and immunohistopathological methods are limited to the assessment of only selected biochemical pathways. In recent years, the use of high-resolution magnetic resonance spectroscopy (MRS) has emerged as a powerful technology for investigating abnormal metabolic compositions, as a wide range of metabolites can be detected simultaneously and “without prejudice” (Table 3⇓). MRS-based metabolomics and metabonomics allow for simultaneous assessment of lipid, carbohydrate, amino acid, and high-energy phosphate metabolism (Figure 1⇓). 1H-MRS analyses of human and animal biosamples––such as tissue biopsies, blood, urine, cerebrospinal fluid, saliva––allow for simultaneous detection of hundreds of low molecular weight (≤20 kDa) metabolites within a biological matrix, at concentrations above 10 μM (73, 80, 82, 83). In addition, using non-radioactive [13C]-labeled tracers in MRS, it is possible to follow uptake and de novo synthesis of glucose, fatty acids, and ketone bodies. Additionally, 31P-MRS allows for quantification of high-energy phosphates, phospholipids, and other phosphor-containing metabolites. One of the biggest advantages of MRS is that the technology can be applied noninvasively in vivo using MRI-MRS scanners––on humans, living animals, perfused organs and cells––therefore, real-time changes in metabolism and their time-dependency can be monitored. Because nearly all major classes of endogenous metabolic intermediates have characteristic MRI spectra, the technique is very useful for fingerprinting disease-related and toxicity-induced metabolic variations.
Analytical Platforms Generating Megavariate Biological Data
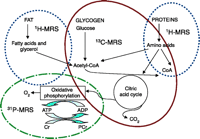
Metabolic pathways amenable to magnetic resonance spectroscopy ( 1 H-, 13 C-, and 31 P-MRS).
Brain Metabolism and MRS
Brain is the organ with the highest demand for oxygen and oxidative energy production. The central intermediate substance of oxidative metabolism is acetyl–coenzyme A (acetyl-CoA), which can originate from carbohydrates, fatty acids, or amino acids (Figure 2⇓). In contrast to other organs, brain is able to utilize glucose almost exclusively to produce ATP; under starvation conditions, ketone bodies can be used as an additional fuel in the brain. Therefore, β-oxidation of fatty acids (e.g., palmitate) to triglycerides, and further, to acetyl-CoA does not occur in the brain, and, as depicted in Figure 2⇓, glucose is the major source for acetyl-CoA production in the brain. Intermediate glucose metabolism takes place mostly in the cytosol through glycogenolysis and glycolysis. Pyruvate, the end-product of cytosolic glycolysis, can be metabolized to lactate by lactate dehydrogenase (LDH), or transported into mitochondria. In the mitochondrion, pyruvate undergoes oxidative decarboxylation to acetyl-CoA, a process catalyzed by pyruvate dehydrogenase (PDH). Acetyl-CoA enters the Krebs cycle by its metabolic conversion to citrate. Alternatively, pyruvate can enter the Krebs cycle through pyruvate carboxylase activity to oxaloacetate by the anapleurotic pathway. The NADH produced in the Krebs cycle is then utilized in oxidative phosphorylation, the major energy production pathway. As a result, one glucose molecule yields thirty-six molecules of ATP: two ATP through glycolysis, two ATP through the Krebs cycle, and thirty-two through oxidative phosphorylation. It should be obvious that disturbances in mitochondrial glucose metabolism would lead to a consider-able decrease in energy production, which, in the brain, cannot be compensated by β-oxidation of fatty acids.
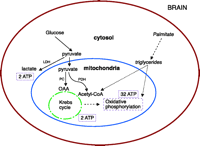
Energy production in the brain. The brain, in contrast to other organs, utilizes only glucose to produce ATP and lacks the enzyme for β-oxidation of fatty acids. Metabolism of one molecule of glucose produces thirty-six molecules ATP: Two ATP through cytosolic glycolysis, two ATP via the mitochondrial Krebs cycle, and thirty-two molecules through mitochondrial oxidative phosphorylation. LDH, lactate dehydrogenase; OAA, oxaloacetate; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase.
Cyclosporine Metabolic Toxicity and Mitochondrial Dysfunction
MRS experiments performed on neuronal cells and on perfused brain slices showed that cerebral energy metabolism is the most sensitive indicator of cyclosporine neurotoxicity in vitro (12–14). Even 100 ng/ml cyclosporine added to incubation medium or perfusate––a concentration well within the range of target cyclosporine trough blood concentrations in transplant patients––reduces high-energy phosphate (ATP) concentrations by 20%. Higher concentrations of cyclosporine (500 ng/ml) produce a decrease of neuronal marker N-acetyl aspartate (NAA) and of neurotransmitter glutamate (12). Astrocyte swelling occurs at cyclosporine concentrations as high as 5000 ng/ml (13). In addition, 13C-MRS monitoring of perfused brain slices demonstrated that in the first four hours of perfusion with 500 ng/ml cyclosporine, production of [4-13C]-labeled glutamate from [1-13C]-labeled glucose was decreased by 40% (84). To compensate for the inhibition of mitochondrial glucose metabolism (a 40%-decrease of NAD+ from oxidative phosphorylation was also observed in 31P-MRS of cyclosporine treated slices), cytosolic glycolysis was increased between four and ten hours of cyclosporine perfusion (a 140% increase of [3-13C]-labeled lactate). Inhibition of mitochondrial glucose metabolism (the Krebs cycle and oxidative phosphorylation) was accompanied by increased reactive oxygen species (ROS) production (17, 84).
The same pattern of metabolic changes was found in rat brain after in vivo treatment with cyclosporine (10 mg/kg/day for 6 days) (16). A decrease in ATP concentrations, accompanied by decreased concentrations of glutamate and g-amino butyric acid (GABA), neurotransmitters generated from the Krebs cycle, and a reduction of NAD+ concentrations were identified by MRS in rat brain extracts. Increased lactate concentrations were found in rat brain as well as in the blood after six days of cyclosporine treatment (16, 73). In addition, increased concentrations of oxaloacetate––the “end” product of the pyruvate metabolism cycle through pyruvate dehydrogenase, or the “first” product of pyruvate metabolism through pyruvate carboxylase––were observed in brain, suggesting that cyclosporine inhibits the Krebs cycle at the very beginning, most likely by reducing PDH activity (Figure 2⇑).
Our findings in rat brain were confirmed in experimental studies on isolated rat kidney mitochondria (85). Exposure of kidney mitochondria to cyclosporine treatment led to a decrease of both succinate- and, especially, glutamate+malate–supported respiration in a dose-related manner. We found that increased lactate production indicated that the negative effects of cyclosporine on mitochondrial metabolism were being compensated by increased generation of ATP via anaerobic glycolysis. In the peripheral blood of renal transplant recipients LDH activity increased just one day after cyclosporine treatment and remained high during the treatment period (30 days) (86). However, succinate dehydrogenase (SDH) activity did not change. Another study in skeletal muscle cells confirmed increased activity of LDH after cyclosporine treatment, and, at the same time, decreased pyruvate dehydrogenase (PDH) activity, which was related to calcineurin inhibition by cyclosporine (87).
Cyclosporine Neuroprotection in Ischemia–Reperfusion
Simultaneously with cyclosporine-mediated neurotoxicity, cyclosporine ameliorates brain injury induced by cerebral I/R in vivo (10, 88). The proposed mechanism of neuroprotection of cyclosporine involves its ability to regulate the mitochondrial permeability transition (MPT) pore (11). Cyclosporine inhibits the binding of the mitochondrial matrix-specific cyclophilin D to the adenine nucleotide translocase in the inner membrane of mitochondria, which is a part of the MPT pore (89). By inhibiting the binding of intramitochondrial cyclophilin D, cyclosporine reduces mitochondrial permeability transition, which is normally triggered by Ca2+-overload of mitochondria and metabolic changes during I/R such as low pH and high concentrations of ADP (Figure 3⇓). Cyclosporine-related neuroprotection is observed in both ischemia and following reperfusion; however, it is suggested that development of MPT exists only in reperfusion phase of I/R (89).
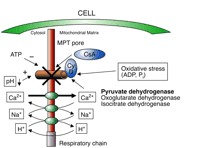
Factors regulating mitochondrial permeability transition (MPT) pore opening. Oxidative stress, an increase in ADP, low pH, or Ca2+ over-load in the mitochondrion during I/R injury can lead to the opening of the MPT pore. Cyclophilin D (Cy D) is an active part of the MPT pore; binding of cyclosporine (CsA) to its immunophilin can prevent the MPT pore opening. Proposed mechanisms in normoxia: cyclosporine increases intramitochondrial Ca2+ (probably by binding calcineurin), increases oxidative stress, and decreases oxidative phosphorylation and activity of the Krebs cycle enzymes (pyruvate dehydrogenase, oxoglutarate dehydrogenase and isocitrate dehydrogenase, as shown in the figure). Cyclosporine mimics hypoxia by derailing respiratory chain, increasing ROS, and decreasing ATP and triggers an opening of the MPT pore ⇒ mitochondrial toxicity. Proposed mechanisms in I/R injury: cyclosporine binds to cyclophilin D and inhibits MPT pore opening triggered by external oxidative stress ⇒ protection. Interestingly, cyclosporine limits its own toxicity by inhibiting these very same effects through inhibition of cyclophilin D. Cyclosporine inhibits the MPR pore opening and therefore antagonizes its own effects on ATP deterioration and ROS production.
We studied the effects of cyclosporine on brain metabolism in perfused rat brain slices using ex vivo MRS under normoxic and hypoxic conditions and during reperfusion. Our studies showed a significant overlap between cyclosporine concentrations inhibiting ATP production in normoxia and those protecting the energy state during hypoxia. For example, cyclosporine concentration of 500 ng/ml significantly decreased brain ATP concentrations up to 20% in normoxia when the slices were perfused with oxygenated medium (14, 15, 17). During subsequent hypoxia in the same slices, the decrease of ATP was 2.4-fold in the presence of cyclosporine in comparison to 9.1-fold in untreated slices (Figure 4⇓) (17). In addition, a systematic dose-finding study showed that the concentration of 500 ng/ml cyclosporine was most efficient for protecting intracellular Ca2+ homeostasis during hypoxia (17).
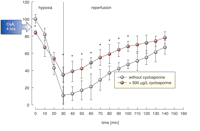
Time-dependent changes of high-energy phosphate concentrations in rat brain slices during hypoxia and reperfusion as continuously monitored by31P-MRS. Cyclosporine-treated slices (open circles) were pre-treated with 500 μg/l cyclosporine for 4 hours. Data are mean ± standard deviation (n= 5). *, p < 0.05.
In contrast to the belief that low cyclosporine concentrations are rather neuroprotective whereas high cyclosporine concentrations are rather neurotoxic (88, 90), our study indicated a significant overlap of the cyclosporine concentrations that protect brain energy metabolism from I/R injury and the concentrations that result in significantly reduced brain energy metabolism under normoxic conditions. Under normoxic conditions, inhibition of mitochondrial energy metabolism is the primary effect; under hypoxic conditions, the inhibition of Ca2+ efflux and protection of mitochondrial function is the primary effect. Other mechanisms may also significantly contribute to the protective effects of cyclosporine during hypoxia. Prior to hypoxia, cyclosporine mimics, to a certain extent, similar metabolic effects of hypoxia and therefore, in hypoxia cyclosporine protects mitochondrial energy metabolism by the effect known as “ischemic preconditioning” (91). The principle of ischemic preconditioning is based on the observation that brief periods of ischemia prior to a major ischemic insult protect tissues against I/R injury. Thus, by inducing metabolic changes similar to hypoxia, cyclosporine exhibits pre-conditioning effects in ischemia and then inhibits MPT opening in I/R.
Cyclosporine Concentrations in The Brain
Is cyclosporine-mediated neurotoxicity caused by a direct effect of cyclosporine on nervous tissue, or by an indirect effect through vasoconstriction? An argument against the direct effect of cyclosporine was based on the findings of earlier studies that cyclosporine cannot cross the blood–brain barrier (92). However, later reports demonstrated that cyclosporine was present in cerebrospinal fluids of patients treated with the drug (7–9). In the brain of lung-transplanted monkeys treated with cyclosporine, low cyclosporine concentrations were found in the brainstem (tissue-to-blood partition coefficient: 0.3), cerebellum (0.2), and cerebrum (0.3) (93). Even though distribution of cyclosporine into the brain was less than its distribution into other target organs of cyclosporine toxicity (i.e., kidney; kidney-to-blood distribution coefficient: 4.3), cyclosporine was detected in the brain of each study animal. Finally, our recent studies (15–17) have shown that in the rat, cyclosporine not only penetrates brain tissue, but that the drug is also found in mitochondria isolated from cyclosporine treated brain, in vivo as well as in vitro as summarized in Table 4⇓.
Cyclosporine Concentrations in Rat Brain, Brain Slices, and Mitochondria
Conclusions
Cyclosporine-dependent neurotoxicity appears in up to 60% of cyclosporine-treated patients, even when cyclosporine blood-trough levels are within the therapeutic range. Clinical appearances of cyclosporine neurotoxicity mimic those of mitochondrial encephalopathies. Mitochondrial dysfunction has widespread deleterious ramifications for cellular function, leading to impaired energy production, impaired cellular calcium buffering, activation of proteases and phospholipases, activation of nitric oxide synthase, and generation of free radicals (75). These pathways can lead to either apoptotic or necrotic cell death depending upon the severity of the insult. Systematic studies using MRS-based metabonomics showed that under normoxic conditions cyclosporine: 1) inhibits the mitochondrial Krebs cycle and subsequently activates glycolysis and lactate accumulation, 2) depletes mitochondrial high-energy phosphate production, and 3) increases production of ROS in the brain––all typical biochemical symptoms observed in patients with mitochondrial encephalopathies (ME). During hypoxia and reperfusion, cyclosporine protects brain energy metabolism, intracellular pH, calcium homeostasis and inhibits production of oxygen radicals. Possible mechanisms include inhibition of cyclophilin D–mediated MPT and ischemic preconditioning. We conclude that cyclosporine’s effect on mitochondria is key to determining neurotoxicity and neuroprotection. Whether the net effect is toxicity or protection depends on the concentrations of cyclosporine and oxygen.
Acknowledgments
Studies in the authors’ laboratories reported here were supported in part by the Alexander von Humboldt Foundation (N.J.S.) and German Research Foundation (N.J.S. and U.C). Preparation of this review was supported in part by the National Institutes of Health (NIH RO1 HL071805-01 and NIH RO1 DK065094-01). We thank Ms. Jaimi Brown for the technical support in manuscript preparation.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Uwe Christians, MD, PhD (bottom), is an Associate Professor and Associate Chair of Research of the Department of Anesthesiology at the University of Colorado Health Sciences Center in Denver. He directs the department’s clinical research programs.

Natalie Serkova, PhD, is an Assistant Professor in the Department of Anesthesiology at the University of Colorado Health Sciences Center in Denver and Director of the Biomedical MRI/MRS facility. She is interested in the development of clinical biomarkers for pharmaco- and toxicodynamic drug monitoring using quantitative metabonomics.

Leslie Z. Benet, PhD, is a Professor in the Department of Biopharmaceutical Sciences at UCSF. He has a long-term interest in the pharmacokinetics and pharmacodynamics of immunosuppressive drugs.

