The gβγ Dimer as a Novel Source of Selectivity in G-Protein Signaling: GGL-ing AT CONVENTION
Abstract
Heterotrimeric G proteins relay information between cell surface receptors and effector molecules in diverse signaling pathways to mediate critical cellular processes in both physiologic and pathologic conditions. Multiple isoforms of each of the three G protein subunits yield enormous structural and functional diversity. G proteins are thus obvious molecular targets for the therapeutic manipulation of signaling pathways. Their ubiquity among a vast array of G protein–coupled receptor pathways, however, may at first seem to threaten the attractiveness of G proteins as drug targets for specific signaling processes; in order for G proteins to be effective targets, some degree of selectivity must be defined and exploited. Although a great deal has been determined about the functional selectivity of Gα subunits, relatively little is known regarding Gβγ selectivity. In this review, we discuss functional diversity among Gβγ subunits in both receptor coupling and effector activation. The novel functions of Gβ5, in complex with proteins of the GGL domain–containing R7 subfamily of regulators of G protein signaling, are discussed in detail, with specific focus on the potential of the Gβ5–RGS9-2 pair as a therapeutic target in Parkinson’s disease.
Introduction
Heterotrimeric guanine nucleotide binding proteins (G proteins) transmit signals across the plasma membrane by undergoing conformational changes, activating enzymes, and participating in multiprotein complexes, thereby allowing cells to transduce diverse external stimuli (e.g., morphine and light) into signals that culminate in neurotransmission, growth, differentiation, or cell death. In the GDPbound basal state, the G protein subunits form an inactive heterotrimer (consisting of Gα, Gβ, and Gγ subunits) that can associate with the cytosolic domains of heptahelical G protein–coupled receptors (GPCRs). In the GTP-bound (i.e., activated) state, the Gα subunit dissociates, and both it and the Gβγ dimer can then interact with effector molecules to initiate signaling cascades [for review, see (1)]. These effectors can be enzymes such as phospholipase C (PLC) or adenylyl cyclase (AC), which produce second messengers that regulate intracellular calcium and cyclic AMP concentrations, respectively, or ion channels, such as the G protein–regulated inwardly rectifying K+ (GIRK/Kir3.0) channels or N-type Ca2+ (Cav2.2) channels.
Regulation of the Gα subunit is determined by nucleotide exchange, wherein dissociation of GDP allows replacement by the more abundant GTP, as well as the subunit’s intrinsic GTPase activity. The exchange reaction is catalyzed by ligand activation of receptors coupled to the GDP-bound heterotrimer; once the ligand binds, the receptor induces a structural change in the α subunit resulting in displacement of GDP (2). Efficient coupling of the heterotrimeric G protein to receptor requires all three subunits (3–8) and causes a conformational change in the receptor, increasing its affinity for ligand (3). Therefore, ligand-induced nucleotide exchange is the culmination of a series of complex interactions among four polypeptides and a ligand (Figure 1⇓).

Standard model of heterotrimeric G-protein signaling. Heterotrimeric guanine nucleotide–binding proteins exist in the basal state as a GDP-bound Gα subunit (red) bound to a Gβγ dimer (blue and yellow). Ligand-bound receptor (green), serving as a guanine nucleotide exchange factor (GEF) for the Gα subunit, catalyzes the release of GDP and the binding of GTP. GTP-bound Gα separates from the Gβγ dimer and thus allows Gα and Gβγ to regulate their respective effectors. The cycle is completed upon hydrolysis of GTP to GDP, resulting in reassociation of the GDP-bound Gα with Gβγ. The Gα subunit has an intrinsic guanine nucleotide triphosphatase (GTPase) activity, which can be stimulated by regulators of G protein signaling (RGS proteins) and, in some cases, the Gα effector.
The GTPase reaction, while intrinsic to the Gα subunit, can be dramatically increased by GTPase-activating proteins, or GAPs (9). A diverse protein family of regulators of G protein signaling (RGS) that act as GAPs for Gα subunits has emerged over the last decade (10–13). By accelerating the return of the activated GTP-bound Gα protein to its basal GDP-bound state, RGS proteins terminate effector activation by both Gα and Gβγ subunits, thus regulating the kinetics and amplitude of signaling.
Gβγ Signaling: Diversity and Selectivity
The requirement for G proteins in the propagation of critical signals across the plasma membrane suggests that pharmacologic manipulation of these proteins may be a lucrative therapeutic endeavor. Further, the diversity of downstream pathways and cellular responses suggests that G proteins must exhibit selectivity in their signaling coupling; such selectivity, moreover, is essential to therapeutic intervention. Gβγ dimers provide a great potential for diversity and selectivity. There are five genes for Gβ and at least twelve for Gγ (Figure 2⇓), yielding an enormous diversity of potential dimer combinations. Most of the combinations can form dimers (14); however, there are exceptions, such as the Gβ2 protein that can pair with Gγ2 but not Gγ1 (15). Nevertheless, the numerous possible Gβγ dimer combinations suggest functional selectivity. Selectivity could occur in at least four different levels: at the G protein–receptor interface, at the G protein–effector interface, via other unidentified Gβγ-interacting cellular components, or due to post-translational modification (e.g., C-terminal lipidation).
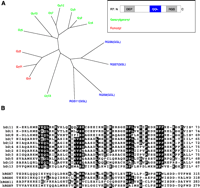
The relationship between the human Gγ subunit proteins and R7 subfamily of RGS proteins. A. An unrooted dendrogram depicts the degrees of similarity among proteins. As shown in the inset, R7 subfamily members contain a DEP domain, GGL domain, and RGS domain; only the GGL domain (blue) sequences were used for the dendrogram analysis. Gγ subunits are post-translationally modified with a geranylgeranyl (green) lipid or a farnesyl (red) lipid moiety. The unrooted dendrogram was generated using TreeView X from a multiple sequence alignment created with ClustalW using default settings. B. Multiple sequence alignment of human Gγ subunits and GGL domains of human R7 subfamily of RGS proteins. Black boxes depict identical amino acids shared by at least 60% of sequences within alignment. Sequence alignment between human Gγ subunits and GGL domains was computed by the Wisconsin GCG Pileup program using default parameters. GenBankTM gi identifier numbers for human sequences used: RGS7, 17380284; RGS6, 31742476; RGS9, 8475983; RGS11, 34452688; γ11, 4758448; γ1, 11386179; γ8, 3023844; γ7, 4826746; γ12, 40254926; γ4, 4758450; γ2, 11277005; γ3, 6912394; γ5, 4885287; γ10, 4758446; γ13, 7706567.
Unfortunately, much of the literature reports overlapping functionality (14,16), perhaps reflecting technical limitations: The Gβ and Gγ subunits cannot be expressed independently, making observations of independent Gβ or Gγ function difficult. In addition, exogenous overexpression of Gβγ heterodimer can potentially overwhelm cellular components that may participate in dimer selectivity. Reconstitution assays can sometimes demonstrate quantitative differences in receptor coupling or effector regulation in vitro but cannot assess the importance of additional cellular components potentially necessary for selectivity. Nevertheless, a number of laboratories have investigated differences in Gβγ specificity at multiple levels (17, 18) and recent studies have demonstrated exquisite subunit selectivity among Gβ and Gγ subunits at both the receptor–G protein interface and the G protein–effector interface.
Gβγ specificity in receptor coupling
Coupling of Gαs-coupled receptors to G protein heterotrimers appears to be influenced by Gβγ subunit composition. Using purified recombinant Gβγ dimers containing the γ2 and the β1, β2, β3, β4, or β5 subunits, McIntire and colleagues measured EC50 values for nucleotide exchange induced by ligand binding to β1-adrenergic receptors (19). Whereas dimers containing β1, β2, β3, or β4 subunits (i.e., Gβ1-4γ2) tend to display similar EC50 values (0.5–2.7 nM), the Gβ5γ2 dimer coupled poorly (17.1 nM). Clearer differences were detected when the investigators examined the A2a adenosine receptor: The most efficient coupling occurred with Gβ4γ2 (1.3 nM), and the least efficient occurred with Gβ5γ2 (232 nM); Gβ1γ2 coupled poorly (15.7 nM), whereas Gβ2γ2 (5.7 nM) and Gβ3γ3 (5.9 nM) coupled with moderate efficiency. The poor ability of Gβ5γ2 to couple to the Gαs-coupled β1-adrenergic and A2a receptors is likely indicative of its selectivity for Gαq-coupled receptors (20).
Compared to the Gβ subunit, the Gγ exists in a more extensive array of isoforms (Figure 2⇑), suggesting a potentially greater role in Gβγ dimer selectivity. The suppression of Gγ7 expression in HEK 293 cells results in the coincident degradation of Gβ1 but not Gβ2–5 monomers (21); prostaglandin E1-induced adenylyl cyclase activity and carbachol- and ATP-induced phosphoinositide turnover were unaffected. Isoproterenol-induced adenylyl cyclase activity, however, was abated, suggesting a specific role for Gγ7 in β-adrenergic receptor signaling in HEK 293 cells (22). These studies led Robishaw’s group to generate Gγ7 knockout mice, which, although fertile and of normal weight, demonstrate an increased startle response (23). Most notably, Gαolf expression in the striatum is reduced 82%, whereas Gαs, Gαo, Gα13, and Gαq are expressed normally. The mice also exhibit reduced D1 dopamine receptor-induced adenylyl cyclase activity specifically in the striatum, mimicking the reduced D1 receptor- induced adenylyl cyclase activity demonstrated previously in Gγ7-suppressed HEK 293 cells (24). Taken together, these results indicate specific functions for Gγ7: Gβ1γ7 dimers couple to β-adrenergic receptors in HEK 293 cells to mediate activation of adenylyl cyclase activity; the Gγ7 subunit also appears to couple with Gαolf and the D1 dopamine receptor to regulate adenylyl cyclase activity in the striatum. Further studies are required to determine if selective functions exist for additional Gγ subunits.
Gβγ Specificity in Effector Activation
In addition to demonstrating a role for Gβ in receptor coupling specificity as described above, McIntire et al. also examined the ability of the various Gβγ2 dimers to activate adenylyl cyclase II (ACII) and inhibit adenylyl cyclase I (ACI) (19). The Gβ5γ2 dimer activated ACII poorly (EC50 =76 nM) and had no measurable effect on ACI, whereas Gβ1γ2, Gβ3γ2, and Gβ4γ2 activated ACII with EC50values of 3.5–5.5 nM. In contrast, Gβ2γ2 activated ACII relatively poorly (EC50 = 12.9 nM). Adenylyl cyclase I was inhibited equally well by Gβ1γ2, Gβ2γ2 and Gβ4γ2 (IC50 = 10.5–16.8 nM) but was inhibited very poorly by Gβ3γ2 (IC50 = 110 nM). Thus, Gβγ dimers containing various Gβ subunits differentially interact with adenylyl cyclase effector enzymes.
More recently, Paula Barrett’s laboratory has demonstrated that the Gβ2 isoform inhibits the low-voltage-activated (LVA) T-type calcium channel α1H (Cav3.2) with exquisite selectivity (25). This inhibition does not appear to depend on any particular geranylgeranylated Gγ isoform; however, Gγ11 fails to inhibit the channel when paired with Gβ2, which may reflect the farsenylation of Gγ11 (Figure 2⇑). Interestingly, the α1G channel (Cav3.1) does not exhibit this Gβγ dimer selectivity, demonstrating differential regulation among effector isoforms, a useful feature for potential pharmacologic targeting.
Although differences in Gβγ signaling may be a direct result of G protein–receptor and G protein–effector interactions, it is likely that a number of in vivo cellular factors also play a role. Diversé-Pierluissi and colleagues have measured the inhibition of N-type Ca2+ currents that ensues when endogenous PLC-β is activated following the introduction of recombinant Gβ1-containing, but not Gβ2-containing, dimers into avian sensory neurons (26). Surprisingly, such selective activation of PLC-β is not seen using in vitro assays, suggesting that additional cellular factors play a role in Gβγ dimer selectivity.
Gβγ Signaling: Novel Aspects of the Gβ3and Gβ5 Subunits
The Gβ3 Subunit
Alternative splicing of the five genes that encode Gβ subunits introduces even greater potential for the functional diversity of Gβγ dimers. A novel splice variant of Gβ3, resulting from a single nucleotide polymorphism (i.e., C825T), has been associated with essential hypertension. The alternative splicing results in the deletion of nucleotides 498–620 from exon 9, putatively resulting in a six-bladed propeller structure, instead of the seven-bladed wild-type Gβ3 protein (Figure 3⇓) (27). The shorter Gβ3 protein, Gβ3s, is biologically active and likely increases signals generated from pertussis toxin–sensitive G proteins (i.e., Gi/o proteins) (27). This increased Gi/o signal transduction is believed to increase smooth muscle cell proliferation and hypertrophy, resulting in an increased susceptibility to hypertension. Gβ3s has also been associated with increased susceptibility to ischemic stroke in Caucasian patients (28) and left ventricular hypertrophy (29).
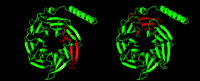
Deleted regions of variant Gβ3subunits. The Gβ1 structure (PDB accession number 1TBG) is depicted, with sequences homologous to Gβ3 shown in green; sequences that are deleted to result in shorter Gβ3 variants are shown in red. Left, Gβ3s; right, Gβ3s2.
The increased proliferation of smooth muscle cells may also contribute to the arteriolar hypertrophy and sclerotic lesions seen in chronic kidney allograft rejection (28), and a link between Gβ3s expression in donor kidneys and an increased susceptibility to tissue rejection has been observed. Neutrophils that contain the gene for Gβ3s manifest an enhanced chemotactic response to N-formyl-Met- Leu-Phe (fMLP) receptor activation (30), and chronic lymphocytic leukemia (B-CLL) patients containing this polymorphism show an increased relapse rate (31). Thus, immune system function may be altered in patients expressing the Gβ3s protein.
More recently, another biologically active polymorphism in the gene for Gβ3, resulting in Gβ3s2, a putative six-bladed protein structure (Figure 3⇑), appears to associate with expression of the C825T polymorphism (32). The relative contributions of Gβ3s and Gβ3s2 to the above disease processes, as well as the mechanisms underlying these contributions, remain to be determined.
The Gβ5 Subunit
Gβ5: A Divergent, Brain-Enriched Subunit
The Gβ5 subunit is an outlier in the Gβ protein family (Figure 4⇓), with only 50% identity to the four other family members, which among themselves share greater than 80% sequence identity (33, 34). The gene for Gβ5 was originally cloned in 1994 by Watson et al. from a mouse brain library; northern blots of various mouse tissues showed robust mRNA expression in the brain and considerably lower expression in the kidney, heart, lung, and skeletal muscle (33, 34). Shortly after Gβ5 was cloned, a retinal-specific, longer splice variant, Gβ5L, was discovered (35). This longer form contains a 42-residue N-terminal extension. Although the shorter Gβ5 subunit (Gβ5S) is also expressed in the retina, only the longer Gβ5L is expressed in the rhodopsin-rich rod outer segment membranes, suggesting an involvement of Gβ5L in phototransduction (see below) (35). The human Gβ5 gene is highly similar to the mouse form (99.4% protein sequence identity), and is also expressed primarily in brain and retina (Figure 4⇓) (34, 36–38). In the brain, the Gβ5 subunit is associated with the membrane; however, a significant amount (30%–50%) has also been found in the soluble fraction (35, 38, 39), which is in contrast to other Gβ subunits which are generally only found associated with the membrane (14, 45, 38).
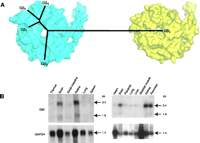
The novel nature of the Gβ5subunit. A. An unrooted dendrogram depicting the relationship among human Gβ subunit protein sequences is superimposed over a space-filling model structure of the Gβ1 subunit (cyan; PDB id 1TBG) and a space-filling model of the Gβ5 subunit (yellow; model coordinates generously provided by Dr. John E. Sondek, UNC-Chapel Hill). B. Northern blot analyses of human Gβ5 transcript expression patterns. Blots of 20 mg total RNA (left) or 2 μg poly(A+) RNA from various human tissues were serially hybridized with a mouse Gβ5 cDNA probe and, as a control for RNA loading and quality, a human glyceraldehyde-3-phosphate dehydrogenase probe. Data adapted from Snow et al. (1998) Proc. Natl Acad. Sci. USA, 95, 13307–13312. Copyright ©1998 by The National Academy of Sciences of the United States of America.
The Gβ5Subunit and RGS Proteins
The divergence of the Gβ5 from the other Gβ subunits, along with its relative preponderance in the soluble cell fraction, may suggest novel binding partners (35, 38). Accordingly, Cabrera et al. investigated the existence of soluble factors that may interact with Gβ5, and chromatographically observed a ~55-kDa protein, later identified as RGS7, that coelutes with soluble Gβ5 (39). Similar approaches have isolated in vivo heterodimers of Gβ5 with the related proteins RGS9 (40) and RGS6 (41).
RGS6, RGS7, RGS9, and RGS11 are members of the R7 subfamily of proteins containing an RGS domain that accelerates the intrinsic GTPase activity of Gα subunits, thereby promoting the formation of inactive G protein heterotrimers and terminating signaling from Gα subunits and Gβγ dimers (42–44). In addition to the RGS domain, R7 proteins contain a Dishevelled/Egl-10/Pleckstrin (DEP) homology domain that mediates membrane association (45, 46) and a G protein gamma-like (GGL) domain that is responsible for dimerization with Gβ5 (36, 47, 48).
Gβ5/R7 Dimerization
The highly specific association of Gβ5 with the GGL domain of R7 family subunits is required for the stability of both members of the heterodimer. Deletion of the GGL domain from RGS7 eradicates the ability to bind Gβ5 (48); conversely, truncated mutant proteins that only contain the GGL and RGS domains of RGS6, RGS7, or RGS11 competently associate with Gβ5 (36, 47). The interaction with GGLcontaining RGS proteins is specific to the Gβ5 subunit; Gβ1, Gβ2, Gβ3 and Gβ4 are unable to associate with RGS6, RGS7, or RGS11 (36, 47, 49). However, exchanging the GGL domain of RGS7 with a portion of the sequence from Gγ1 allows for association with Gβ1 (48). Because of the considerable amino acid sequence homology between GGL domains and Gγ subunits (Figure 2⇑), it is likely that Gβ5 interacts with RGS proteins in a manner structurally analogous to a Gβ–Gγ interaction (50). We have shown that mutation of residues in the GGL domain analogous to residues that are involved in Gβ1–Gγ1 binding diminish or eradicate Gβ5–RGS protein association (47). It is thus unlikely that Gβ5 could simultaneously associate with both an RGS protein and a Gγ subunit (see also Box 1).
The Case For a Gβ5γ Dimer?
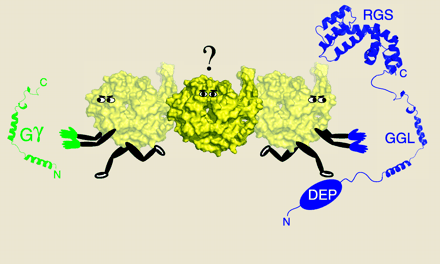
A conundrum currently exists in the Gβ5 field. It is well established that Gβ5–RGS protein complexes exist in vivo (39, 41, 51); however, there is currently no direct evidence for the in vivo existence of Gβ5γ dimers. In fact, some have speculated that Gβ5 only exists in a complex with GGL domain–containing RGS proteins and not with Gγ subunits (50, 51, 85).
Does Gβ5 function in a traditional Gβγ sense? Two general functions characterize all Gβγ dimers: 1) the ability to support ligand-induced, receptor-catalyzed nucleotide exchange on Gα subunits; and 2) the ability to directly regulate effectors (14). Numerous laboratories have examined the ability of Gβ5 to regulate known Gβγ effectors. For example, when Gβ5 was cotransfected with a Gγ subunit (e.g., Gγ2) and PLC-β2 into COS-7 cells, a significant increase in IP3 was observed (33, 35). In comparing Gβ1 and Gβ5 functions in cotransfection assays, Zhang et al. demonstrated that Gβ5γ2 could activate PLC-β2 similarly to Gβ1γ2; however, unlike Gβ1γ2, Gβ5γ2 had no effect on downstream activation of MAPK or c-Jun N-terminal kinase pathways (86). Using purified proteins reconstituted in lipid vesicles, Lindorfer et al. showed that Gβ5γ2 could directly stimulate turkey erythrocyte PLC-β (87). In similar in vitro studies, Maier et al. and Yoshikawa et al. confirmed that the PLC-β2 isoform could be directly activated by Gβ5γ2 (88, 89).
Phosphatidylinositol-3-kinase (PI3K) is another effector known to be regulated by Gβγ dimers (90, 91, 92). Using purified proteins in an in vitro assay system, Maier et al. discovered that Gβ5γ2 has no measurable effect on the enzymatic activity of either PI3Kβ or PI3Kγ (88). Activation of PI3K is known to result in downstream activation of MAPK and c-Jun N-terminal kinase pathways (93–95). The inability of Gβ5γ2 to activate PI3K may be the reason why co-transfection of Gβ5γ2 is unable to activate MAPK and c-Jun N-terminal kinase (86, 88).
On the other hand, Gβ5γ dimers can regulate the function of two brain-enriched effectors. Zhou et al. investigated the ability of different Gβγ dimers to regulate N-type Ca2+ channels by transfecting specific Gβ and Gγ subunits into HEK293 cells stably expressing N-type Ca2+ channels; Ca2+ currents were measured by a whole-cell patch technique (54). The Gβ5γ2 dimer inhibited the channels similarly to other Gγ2- containing dimmers. Most Gβγ dimers that have been examined activate G protein-activated inwardly rectifying K+ channels (96). Gβ5-containing Gβγ dimers, however, inhibit both basal and receptor-activated currents generated by these channels (96). Binding studies demonstrated that Gβ5γ2 can bind to the same channel domains as other Gβγ dimers (96); results from competition binding assays and activity assays suggest that the mechanism of GIRK channel inhibition is competition between Gβ5γ dimers and other Gβγ dimers for GIRK channel interactions (96, 97).
Gβ5γ dimers can also bind Gα subunits and couple to receptors. Fletcher et al. demonstrated that purified Gβ5γ2 could bind Gαq but not Gαi1, Gαi2, Gαo, or Gαs (20). Gαq, and possibly related subunits such as Gα11, can be selectively purified from bovine brain membrane extracts using Gβ5γ2 immobilized on an agarose column (20). Purified Gαq and purified Gβ5γ2 are able to couple to the M1 muscarinic receptor and the endothelin B (ETB) receptor and participate in ligandinduced nucleotide exchange on the Gα subunit (87).
Thus, it appears that the Gβ5 subunit may have two independent binding partners (i.e., Gγ subunits and GGL-containing RGS proteins), and different functions have been described for each complex. Gβ5γ2 can participate in functions that typify Gβγ dimers: functional interactions with a Gα subunit and receptor as well as the ability to regulate known effectors. Unfortunately, there is a dearth of direct evidence to support the in vivo existence of Gβ5γ dimers, which has led some to doubt the relevance of Gβ5γ dimers (50, 85).
Indeed, a number of technical considerations have hampered the experimental investigation of Gβ5γ dimer function. Gβ5 clearly does not engage in the typical Gβ–Gγ dimer interaction. Certain non-denaturing buffer conditions (e.g., ionic detergents such as cholate) appear to perturb the Gβ5–Gγ association (20, 47, 89, 98). Even coimmunoprecipitation procedures have been reported to dissociate Gβ5 from Gγ from lysates known to contain active dimer (99), although the Gβ5–RGS interaction is maintained under similar conditions. Therefore, investigation of Gβ5γ dimers must give due regard to the sensitivity of the Gβ5–γ interaction to experimental conditions.
Gβ5 and R7 proteins are obligate binding partners; expression and solubility of either is limited by stoichiometric expression of the binding partner (39, 40, 51). Data from Gβ5 knockout mice clearly show that RGS6, RGS7, RGS9, and RGS11 protein stability in vivo is also dependent on Gβ5 expression (52). Protein expression of all four GGL-containing RGS proteins is eliminated or greatly reduced in the retina and striatum in the absence of Gβ5. Likewise, in the RGS9 knockout mice, expression of the retinal specific Gβ5L isoform is lost (53). Expression of the shorter Gβ5 isoform is maintained in striatum of RGS9-deficient mice, possibly reflecting an association of Gβ5 with other R7 family members.
GGL/Gβ5Dimers as Conventional Gβγ Pairs
Because GGL-containing RGS proteins associate with Gβ subunits in a manner analogous to the Gγ–Gβ association, one might speculate that Gβ5–RGS complexes play a role in cellular signaling analogous to Gβγ dimers. If Gβ5–R7 dimers promote either receptor–G protein coupling or effector activation (in addition to their GAP function), they would be positioned to regulate G protein activity at multiple levels (Figure 5⇓). To date, no such Gβ5–R7 function has been demonstrated biochemically. In fact, Posner et al. report that recombinant Gβ5–RGS6 and Gβ5–RGS7 dimers do not form heterotrimeric complexes with Gαo•GDP or Gαi•GDP, nor were they capable of directly regulating effectors such as ACI, ACII, ACV, PLC-β1 or PLC- β2 (49). In addition, Gβ5–RGS11 was found to be unable to regulate N-type Ca2+ channels (54).

A model depicting a possible Gα–GDP–Gβ5–R7 heterotrimer coupled to a GPCR. GDP-bound Gα would undergo ligand-induced nucleotide exchange when coupled with a Gβ5–R7 dimer. The Gβ5–R7 complex would be tethered to the membrane via binding of R9AP (or an R9AP-like molecule) to the DEP domain of the RGS protein. Upon activation, the RGS domain of the Gβ5–R7 complex would be conveniently localized to accelerate the hydrolysis of GTP to GDP.
In spite of the lack of biochemical evidence, there is genetic evidence for Gβ5–R7 dimers interacting with GDP–Gα in a Gβγ- like interaction, so as to regulate G protein signaling in nematode worms (13, 55). In C. elegans, the Gαo homolog GOA-1 and the Gαq homolog EGL-30 have opposing effects on locomotion and egglaying behaviors (13, 56). Genetic evidence suggests that the Gαo protein opposes the activity of Gαq via EAT-16, a GGL-containing RGS protein that is a negative regulator of EGL-30 and acts down- stream of Gα signaling (57). There is also evidence that a second GGL-containing RGS protein in C. elegans, EGL-10, is a negative regulator of Gαo (13). The inhibition of both Gαq by EAT-16 and Gαo by EGL-10 are dependent on the expression of the C. elegans Gβ5 homolog GPB-2 (58, 59). Based on these observations, a model has been proposed in which EAT-16 (i.e., the GGL-containing RGS protein) forms a heterotrimer with GDP-bound GOA-1 (i.e., the Gαo homolog) and GPB-2 (i.e., the Gβ5 homolog) (55). Upon activation of GOA-1–coupled receptors, the EAT-16–GPB-2 (i.e., Gβγ-like) heterodimer is freed to serve as a GAP and turn off signaling mediated by EGL-30 (i.e., the Gαq homolog). An attractive implication of the model is the reciprocal inhibition between GOA-1– and EGL-30–coupled receptor activation: the Gβγ-like EGL-10–GPB-2 dimer would form a heterotrimer with EGL-30 and, when released by EGL-30–coupled receptor activation, would act as a GAP to turn off GOA-1 signaling. This model is feasible because, whereas the GGL domain of EAT-16 and EGL-10 interacts in 1:1 stoichiometry with GPB-2, the RGS domain is catalytic and thus may affect multiple Gα subunits. Thus, in the C. elegans model above, activation of receptors coupled via an R7–GPB-2 dimer to one Gα could very efficiently silence signaling from another Gα subunit.
R7 GAP Activity
Several laboratories, including our own, have found the GAP activity of R7 proteins to be specific for members of the Gαi/o family, with the highest GAP activity exhibited toward the brain-enriched isoform Gαo. Shuey et al. demonstrated that the RGS domain in vitro can increase the GTPase activity of soluble purified Gαi1 and Gαo but not of Gαs (60). We have shown that Gβ5–RGS11 dimers in vitro can increase the GTPase activity of soluble Gαo without significantly affecting Gαi1, Gαs, Gαq, Gαz, Gα12, or Gα13 (36). Posner et al. similarly observed Gαo-specific GAP activity in Gβ5–RGS6 and Gβ5–RGS7 dimers (61).
More recently, we published a comprehensive study examining the ability of purified, recombinant Gβ5–R7 dimers to increase the steady-state GTPase activity of Gαi1, Gαi2, Gαi3, Gαo, Gαq, and Gα11 in the context of receptor-coupled heterotrimers reconstituted (with conventional Gβγ dimers) in proteoliposomes (62). We found that dimers of Gβ5 and RGS6, RGS7, RGS9, or RGS11 exhibited GAP activity against Gαi proteins coupled to M2 muscarinic acetylcholine receptors but not against Gαq or Gα11 proteins coupled to M1 receptors (Figure 6⇓). Further, there were differences in the potencies and efficacies of the various Gβ5–R7 dimers in their abilities to accelerate the GTPase activity within the Gαi family. Gαi subunits achieved two- to fourfold higher maximal GTPase rates in the presence of RGS11 or RGS6 as compared to RGS9 or RGS7 (62). Additionally, Gβ5–RGS9 and Gβ5–RGS11 were more potent (EC50 = 25–80 nM) than Gβ5–RGS6 and Gβ5–RGS7 (EC50 = 150–350 nM) for Gαi1, Gαi2, and Gαi3, while all four Gβ5–R7 dimers exhibited similar potency for Gαo (EC50 = 16–47 nM).
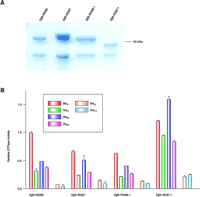
The effects of purified Gβ5–R7 dimers on the GTPase activity of Gi and Gq family Gα subunits. A. Recombinant epitope-tagged Gβ5–R7 dimers were expressed in insect cells and purified; purified products are shown on a coomassie blue-stained SDS-PAGE gel. B. Relative steady-state GTPase activities of various purified Gα subunits reconstituted with receptors in proteoliposomes were determined in the presence of Gβ5–R7 dimers. Gαo, Gαi1, Gαi2, and Gαi3 (with Gβ1γ2) were reconstituted in phospholipid vesicles with the M2 muscarinic receptor, and Gαq and Gα11 (with Gβ1γ2) were reconstituted with the M1 muscarinic receptor (62).
Clearly, the R7 family of RGS proteins exhibits remarkable selectivity for increasing the GTPase activities of Gαi subunits; however, there are numerous reports in the literature of inhibition of Gαq-family signaling pathways by overexpressed R7 family proteins (60, 63–65). It is unclear if the effect on Gαq signaling is an artifact due to R7 overexpression or in vitro conditions. Studies utilizing truncation mutants of R7 family proteins lacking the RGS domain but maintaining the Gβ5-interacting GGL domain will be critical in investigating the possibility of GAP-independent R7 regulation of G protein signaling.
Biology of Gβ5–R7 Dimers
RGS9-1 Regulation of Phototransduction
The most well characterized physiological function of R7 subfamily RGS proteins is the role of RGS9-1, a retinal-specific splice variant of RGS9, in phototransduction in rod and cone cells. Phototransduction is mediated by the photon-activated receptor rhodopsin, which activates the Gi family Gα subunit transducin (Gαt) and, in turn, the Gαt effector cGMP-phosphodiesterase. GTP turnover by Gαt is much too slow (tens of seconds) to account for the fast (<1 s) physiologic inactivation (66, 67). He et al. demonstrated that the recombinant RGS domain of RGS9 (66) or the recombinant Gβ5L–RGS9 complex (68) significantly increases the GTPase activity of Gαt in vitro, suggesting that RGS9-1 is the GAP responsible for the fast inactivation of Gαt. Furthermore, immunodepletion of RGS9 from rod outer segment detergent extracts removes most of the Gαtmodulating GAP activity from the extract (67). Confirming these biochemical data, rod cells from RGS9 knockout mice exhibit slower recovery to light as compared to rod cells from wild-type mice (53).
Two laboratories have recently discovered a small, retinal-specific integral membrane protein that serves as a RGS9 anchor protein (R9AP) (45, 69). R9AP co-purifies with Gβ5L/RGS9-1 from rod outer segment membranes, and the interaction appears to be mediated by the DEP domain of RGS9-1 (46). As the name implies, R9AP functions by anchoring RGS9-1 to the membrane (Figure 5⇑), thereby increasing the effective concentration of RGS9-1 at the membrane of outer segments. This localization enhances the ability of RGS9-1 to increase the GTPase activity of Gαt in the retina (45, 46, 69, 70, 71). In the absence of R9AP, the stability of the Gβ5L/RGS9-1 complex in photoreceptors is severely compromised (72). Studies have yet to show if there are similar anchoring proteins for the other R7 family members or if R9AP similarly anchors other R7 family members. Interestingly, Snapin, a cytosolic homolog of R9AP implicated in SNARE-mediated membrane fusion, binds the DEP region of RGS7 (73) although, in the absence of a membrane targeting function, the significance of this interaction is unknown.
RGS9 and its membrane anchor R9AP have recently been implicated in hereditary abnormalities in photoresponse recovery (70). Patients with recessive mutations that affect either RGS9 or R9AP and result in severely diminished Gαt-directed GAP activity are unable to see moving objects accurately, especially under lowcontrast conditions, and have difficulty adjusting to changes in light intensity. This is the first known human pathology associated with a defect in an RGS protein (70).
RGS9-2 Regulation of Dopaminergic and Opioid Signaling
A second role for the Gβ5–RGS9 dimer in regulating signal transduction is emerging. A longer splice form of the RGS9 gene, RGS9-2, contains a proline-rich 205 amino acid C-terminal extension in addition to the DEP, GGL, and RGS domains. The physiological function of this C-terminal region remains uncharacterized, but recent studies suggest a possible role in substrate selectivity (74) or nuclear localization (75). RGS9-2 expression is highly enriched in striatum, a brain region rich in dopamine and dopaminergic innervation that has been implicated in mediating the reward effects of both dopamine and opioid receptor activation. In addition to its predominant expression in striatum, RGS9-2 is also expressed in the superficial laminae of the dorsal horn, a site implicated in opiate analgesic effects.
Recent in vitro and in vivo work implicates Gβ5–RGS9-2 dimers in the regulation of inhibitory signaling downstream of D2 dopamine receptors (D2Rs) and μ-opioid receptors (μORs), presumably by increasing the GTPase activity of the Gαi subunit. Overexpression of RGS9-2 attenuates D2R-mediated inhibition of adenylyl cyclase activity in HEK293 cells (76) and accelerates the kinetics of D2R-mediated GIRK channel activation in CHO cells (76) and Xenopus oocytes (77). Further, in a melanophore dispersion assay, expression of RGS9-2, but not RGS9-1, lowers maximal responses to μOR agonists (78). Additional studies are necessary to determine if there is specificity for, or direct interaction with, these receptors or if RGS9-2 interacts solely with the activated Gα subunit.
Studies of mice that either overexpress or lack RGS9-2 in striatum support a physiologic role for RGS9-2 in the regulation of dopaminergic and opioid signaling. Viral-mediated overexpression of RGS9-2 specifically in the nucleus accumbens reduces the locomotor response to cocaine and D2R agonists (77). Further, RGS9 knockout mice display heightened locomotor and reward responses to cocaine, and striatal extracts of RGS9 knockout mice show 50% more D2R-mediated inhibition of forskolin-stimulated cyclase activity as compared to wild-type extracts (77). Likewise, RGS9 knockout mice show tenfold higher sensitivity to the reward effect of the opioid receptor agonist morphine as measured in place-preference conditioning assays. This phenotype is reversed by viral-mediated expression of RGS9-2 specifically in the nucleus accumbens of the knockout animals (79). RGS9-deficient mice also show heightened sensitivity to morphine in behavioral assays of analgesic effects (79, 80). These results suggest that inhibition of RGS9-2 may be a valuable adjuvant to opiate-mediated analgesia.
The steady-state level of RGS9-2 protein expression in striatum appears to be regulated by opioid and dopaminergic receptor activation. RGS9-2 protein levels (but not mRNA levels) are significantly increased following chronic cocaine exposure, consistent with a role for RGS9-2 in the development of tolerance (77). The effects of morphine on RGS9-2 expression are more complex. Acute morphine treatment results in an increase in RGS9-2 expression, but chronic treatment decreases expression. Additionally, RGS9-deficient mice show delayed or reduced development of morphine tolerance and enhanced withdrawal symptoms (79, 80). Dysregulation of RGS9-2 (and Gβ5) expression and/or stability represents a potential mechanism for the development of tolerance and addiction to opioid and dopaminergic agents. Additional studies are needed to determine the significance and therapeutic value of this mechanism.
Dopamine, Parkinson’s Disease, and RGS9-2
Parkinson’s disease is a neurodegenerative disease that manifests motor disturbances as a result of deficient striatal dopaminergic signaling. The primary symptoms of the disease are related to dysregulated motor function, including bradykinesia, muscular rigidity, resting tremor, and abnormal posture. The events leading to the onset of Parkinson’s disease remain a mystery, but the role of dopaminergic signaling in the progression of the disease has been recognized for nearly half a century. In 1960, it was shown that levels of striatal dopamine in patients with Parkinson’s disease were reduced more than ninety percent (81). This deficiency reflects a loss of dopaminergic neurons in the nigrostriatal tract that have cell bodies in the substantia nigra and axonal projections onto cholinergic interneurons in the striatum (Figure 7⇓). Dopaminergic stimulation in the striatum leads to two outflow pathways: a direct pathway mediated by excitatory D1 receptor–expressing neurons and an indirect pathway mediated by inhibitory D2 receptor–expressing neurons that converge in the substantia nigra pars reticulate and medial globus pallidus and provide feedback to the cerebral cortex. Both pathways are dysregulated in Parkinson’s disease and contribute to reduced excitatory input to the cortex.
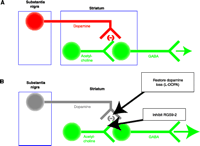
Dopaminergic transmission between the substantia nigra and striatum. (A) Normally, neurons originating from the substantia nigra and terminating in the striatum release dopamine to propagate signals to cholinergic interneurons and GABAergic neurons, leading to inhibition of output from the GABAergic neurons. (B) In Parkinson’s disease, these nigrostriatal dopaminergic neurons are progressively lost (gray), causing an imbalance in the normal control of posture and fine motor movement owing to increased GABAergic output from this circuit. l-DOPA, the immediate precursor of dopamine, is used in anti-Parkinson’s pharmacotherapy to restore dopamine receptor-mediated inhibition of this neuronal circuit. Adding an RGS9-2 inhibitor as an adjuvant to l-DOPA should potentiate dopamine receptor signaling by blunting negative regulation via RGS domain–mediated GAP activity.
Dopamine replacement, using the brain-accessible dopamine precursor l-DOPA, is the cornerstone of therapy for Parkinson’s disease. lDOPA regimens are effective at relieving the motor symptoms in most patients [for review, see (82)]. However, short-term side effects include nausea, vomiting, and cardiac arrhythmia, and long-term treatment may produce dyskinesias, or involuntary movements, and psychiatric disorders (83). Thus, alternate treatment options are necessary. Recent studies of the regulation of dopamine signaling implicate RGS9-2 as a candidate therapeutic target for Parkinson’s disease. In fact, it has been reported that patients with Parkinson’s disease have elevated RGS9-2 immunoreactivity in the postmortem caudate nucleus and putamen, where the increased levels of RGS9-2 correlate inversely to dopamine levels (84). These findings further support a role for RGS9-2 in the progression of the disease and suggest that anti-RGS9 pharmacotherapy might serve as an effective adjuvant to l-DOPA treatment by blunting negative regulation of dopamine receptor signaling within the cholinergic interneurons (Figure 7⇑).
Conclusion
The Gβγ dimer, formerly considered as a mere bystander to the Gα- mediated coupling of GPCRs to effectors, is now known to have its own rich set of downstream signaling targets. Recent evidence has now highlighted that some of these targets are selectively responsive to particular subsets of Gβγ dimers. Moreover, the diversity of potential Gβ–Gγ pairings, of specific expression patterns, and of potential new functions has been complemented by the discovery that Gβ5, by binding to the GGL domains of R7 RGS proteins, is functionally distinct from other Gβ isoforms. Significantly, the emergent properties of the Gβγ dimer should suggest new avenues—be they gene-therapy or small-molecule approaches—to the therapeutic modulation of signaling by GPCRs.
Acknowledgments
The authors would like to thank Dr. T. Kendall Harden for his critical role in developing and supporting the experiments and hypotheses discussed regarding R7 family RGS proteins. SBH is supported by the National Institutes of Health (F32 GM66561). MBJ is a postdoctoral fellow of the Pharmaceutical Research And Manufacturers of America (PhRMA) Foundation. Research on the R7 subfamily of RGS proteins in the Harden and Siderovski labs is supported by NIH grant P01 GM065533.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Miller B. Jones, PhD, is a senior postdoctoral research associate in the Siderovski laboratory. Address correspondence to SBH. E-mail SHooks{at}rx.uga.edu; fax 706-542-5358.

David P. Siderovski, PhD, is an Associate Professor in the Department of Pharmacology at UNC-Chapel Hill and is a member of the UNC Neuroscience Center and the Lineberger Comprehensive Cancer Center.

Shelley B. Hooks, PhD, was a senior postdoctoral research associate in the Harden laboratory at UNC during the preparation of this review article, but is currently on the faculty of the College of Pharmacy at the University of Georgia in Athens.

