Proteases Display Biased Agonism at Protease-Activated Receptors: Location matters!
Abstract
Protease-activated receptors (PARs) are G protein–coupled receptors (GPCRs) that transmit cellular responses begun by the actions of extracellular proteases. The activation of a PAR occurs by a unique mechanism whereby the extracellular N-terminal segment of the inactive receptor undergoes proteolytic cleavage, resulting in irreversible activation––unlike most GPCRs that are reversibly activated. PARs mediate cellular responses to coagulant proteases in various cell types localized within the vasculature. Additionally, PARs are expressed in other cell types and respond to a plethora of proteases. Recent studies have revealed that different proteases elicit distinct responses through the activation of the same PAR. This phenomenon appears to involve stabilization of distinct active PAR conformations that facilitates selectively coupling to different effectors and is localized to caveolae, a subtype of lipid rafts.
Introduction
The four protease-activated receptors (PARs) constitute a family of G protein–coupled receptors (GPCRs) that elicit cellular responses to extracellular proteases. PARs are expressed in many cell types, including cells associated with the vasculature, and are best known for mediating responses to coagulant proteases such as thrombin (1). PARs mediate hemostasis and thrombosis, inflammation, vascular development, and cancer progression (2). In addition to coagulant proteases, several other proteases activate PARs in vitro and may modulate their activity in vivo to regulate various physiological processes in normal and pathological disease states. Specific protease-dependent activation of PARs can differentially regulate cellular responses (3–5) by coupling to multiple effectors. The finding that distinct proteases can differentially activate PAR signaling has added a new level of complexity to PAR signaling. Moreover, the mechanisms responsible for mediating protease-selective PAR signaling are largely unknown.
The observation that different ligands acting at the same receptor can elicit distinct signaling responses has been reported for many GPCRs and is a process termed “functional selectivity” or “biased agonism” (6). The molecular basis of functional selectively involves ligand-induced stabilization of distinct active receptor conformations. New studies indicate that compartmentalization of GPCRs within membrane microdomains also facilitate stabilization of distinct active receptor conformations and promote receptor coupling to specific signaling effectors (7). The unique irreversible mechanism of PAR activation, which results from cleavage of its N-terminal domain at a specific site, suggested that PARs are activated by only one ligand and confounded the concept that proteases were capable of displaying biased agonism at PARs. Our recent study now reveals that compartmentalization of PAR1 in caveolae is critical for protease-selective activation and signaling (5). Here, we discuss PAR activation, signaling, and protease-selective biased agonism.
The Family of PARS
The four PARs, encoded by distinct genes, are a unique class of GPCRs that signal in response to extracellular proteases. The discovery of PAR1 in 1991 resulted from a search for a receptor that conferred thrombin signaling and was originally dubbed the “thrombin receptor” (8). PAR1 is considered the family prototype and is the predominant mediator of thrombin signaling in most cell types. PAR3 and PAR4 were later discovered and also shown to elicit cellular responses to thrombin (9–11). PAR2 was identified in a mouse genomic library screen using probes similar in sequence to the transmembrane regions of the substance K receptor (12). Unlike other PARs, PAR2 is activated by trypsin-like serine proteases but not by thrombin.
Thrombin, the main effector protease of the coagulation cascade, drives fibrin deposition and signals through PARs to promote platelet activation, which is critical for hemostasis and thrombosis (1). Activation of PARs by thrombin also contributes to inflammatory and proliferative responses triggered by vascular injury and thrombotic disease. PARs are expressed primarily in cells of the vasculature, including platelets, immune cells, endothelial cells, fibroblasts, and smooth muscle cells, and exhibit species-specific differences in expression patterns. PAR1 and PAR4 are the functional thrombin receptors present on human platelets (13), whereas PAR3 and PAR4 mediate thrombin signaling in murine platelets (10, 14). In human endothelial cells, PAR1 is predominantly expressed together with PAR3 (15), whereas PAR4 is co-expressed with PAR1 in murine endothelial cells (16). PAR2 is widely distributed and expressed in endothelial cells, fibroblasts, intestinal epithelial cells, and airway cells, mediating inflammatory and proliferative responses associated with tissue injury (17). PAR1 and PAR2 are also expressed in sensory neurons and glial cells and initiate neurogenic inflammation, edema and hyperalgesia, however, the proteases that activate PARs in these particular cell types in vivo have yet to be identified (18). Indeed, with the exception of coagulant proteases and vascular cells, the particular proteases that function as the physiological regulator of PAR activation in a given tissue or cellular setting are not well defined.
Activation and Signaling By PARS
The model for proteolytic activation of PARs posits that proteases cleave a specific peptide bond within the N terminus of the receptor, which results in the formation of a new N terminus that acts like a tethered ligand by binding intramolecularly to the receptor to trigger transmembrane signaling (Figure 1⇓) (8, 19). Synthetic peptides that mimic the tethered ligand sequence of the newly exposed N terminus can activate PARs independent of proteolytic cleavage, with the exception of PAR3 (9). Although typically proteolytic cleavage leads to activation of the same receptor, there is evidence of crosstalk between different PARs. In murine platelets and transfected cells, PAR3 binds to and localizes thrombin to facilitate activation of PAR4, a low affinity thrombin receptor (20). PAR3 is efficiently cleaved by thrombin, but is less efficacious than other PARs at eliciting G protein–dependent cellular responses in vascular cells. Recent work indicates, however, that activation of PAR3 by thrombin induces rapid Ca2+-dependent release of ATP from lung epithelial A549 cells, a cell line that does not express detectable PAR1 or PAR4 (21).

Proteases and peptide agonists display biased agonism at protease-activated receptor 1 (PAR1). PAR1 is a seven-transmembrane G protein–coupled receptor activated by a unique proteolytic mechanism. Thrombin (α-Th), a serine protease, binds to and proteolytically cleaves the N terminus of PAR1, unmasking a new N terminal domain that then acts as a tethered ligand, binding intramolecularly to the body of the receptor to trigger transmembrane signaling. Thrombin-activated PAR1 preferentially couples to G12/13 proteins, which signal to RhoGEFs and result in RhoA activation and endothelial permeability. Synthetic peptide agonists that mimic the newly exposed N terminus–tethered ligand domain, SFLLRN, can activate PAR1 independently of proteolysis. Activation of PAR1 by addition of agonist peptides, SFLLRN or TFLLRNPNDK, promotes preferential coupling to Gq over G12/13. Activated protein C (APC), an anti-coagulant serine protease, requires endothelial protein C receptor (EPCR) as a co-factor and cleaves and activates PAR1. However, in contrast to thrombin, activation of PAR1 by APC promotes endothelial barrier protective signaling predominantly through Gi coupling and is dependent on caveolae, a subtype of lipid rafts.
Formation of heterodimers
In addition, PAR3 dimerizes with PAR1 and consequently potentiates thrombin signaling in endothelial cells, suggesting that PAR3 functions as an allosteric modulator of PAR1 signaling in certain cell types (22). Another type of PAR crosstalk occurs in endothelial cells, where the tethered ligand domain of signaling-defective cleaved PAR1 containing a mutation in a critical proline residue in the second extracellular loop region important for receptor activation, transactivates PAR2 (15), although the physiological significance of this type of intermolecular transactivation remained elusive. During the progression of sepsis, a systemic inflammatory condition characterized by disseminated intravascular coagulation, activated PAR1 switches from endothelial-disruptive signaling (Box 1) to protective signaling via transactivation of PAR2, a receptor whose expression increases in endothelial cells during severe sepsis (23). Thus, the formation of heterodimeric complexes between PARs appears to modulate certain signaling responses but is unlikely to be critical for monomeric PAR coupling to heterotrimeric G-protein signaling (24). Distinct PAR dimeric complexes, however, might have other functions such as facilitating distinct protease-selective signaling, but this remains to be determined.
Endothelial Barrier Integrity and Permeability
Vascular endothelial cells line blood vessels and maintain blood fluidity and endothelial barrier integrity. Most resting endothelial cells in non-injured tissues form tight junctions creating an endothelial barrier, which prevents plasma proteins from moving into interstitial tissues. The formation of the endothelial barrier is mediated by adherens junctions, which facilitate stable cell-cell interactions. The disruption of the endothelial barrier involves intracellular signaling events that lead to disassembly of adherens junctions and reorganization of the actin cytoskeleton and dissolution or loosening of cell-cell interactions. This results in an increase in endothelial barrier permeability. Restoration of endothelial barrier involves a different type of intracellular signaling that promotes re-formation of adherens junctions and formation of tight cell-cell interactions.
Signaling effectors
Once activated, PARs undergo conformational changes that facilitate their coupling to heterotrimeric G proteins. Activated PAR1 couples to Gq, Gi and G12,13, and promotes diverse cellular responses (Figure 1⇑). Several early studies indicated that PAR1 accomplishes this by inhibiting cAMP accumulation through Gi and stimulating phospholipase C (PLC)-catalyzed hydrolysis of phosphoinositides (PI) and Ca2+ mobilization through Gq (25–27), whereas activating the extracellular signal-regulated protein kinases 1,2 (ERK1,2) occurs through both Gq and Gi signaling (28). Other studies have illustrated coupling of PAR1 to G12/13, which leads to activation of Rho GEFs, induction of cytoskeletal changes, and PLC activation (29–31). PAR4 also appears to couple to G q and G12/13 but not Gi. Previous studies questioned whether PAR3 is capable of signaling autonomously (20); however, new work indicates that PAR3 can elicit thrombin-dependent cellular responses in particular cell types (21). Although there is no direct evidence linking PAR2 to heterotrimeric G-protein activation, numerous studies demonstrate that activation of PAR2 with proteases and/or synthetic peptide agonists increase second messenger responses suggestive of Gq, Gi and perhaps G12/13 signaling. Activated PAR2 also binds to and internalize with β-arrestin, a multifunctional adaptor protein (32, 33). Once internalized, the PAR2-β-arrestin complex functions as a scaffold to recruit ERK1,2 on endocytic vesicles and is thought to sustain ERK1,2 signaling in the cytoplasm independently of G-protein activation. The activation of distinct G-protein subtypes as well as non-G-protein effectors by PARs is crucial for eliciting cell type-specific responses. The extent to which PARs couples to distinct G-protein subtypes in a particular cell type depends in part on the G-protein and effector repertoire expressed in the cells but other mechanisms are likely to exist.
Multiple proteases activate PAR signaling
In addition to thrombin, other proteases activate PAR1. Tissue factor (TF), a single transmembrane protein, initiates coagulation by generating thrombin through activation of coagulation factor (F) VIIa and FXa and also promotes cellular signaling via activation of PARs. The upstream coagulation protease FXa can, by itself, proteolytically cleave and directly activate PAR1 or do so in a complex with TF and FVIIa (34, 35). Activated protein C (APC), when bound to endothelial protein C receptor (EPCR), a single spanning transmembrane protein, cleaves and inactivates FVa and VIIa to diminish thrombin generation and induces cellular responses through the activation of PAR1 (Figure 1⇑) (36). APC, an anticoagulant protease, is generated on the endothelial cell surface via activation of protein C (PC) by the complex composed of thrombin and thrombomodulin, an integral membrane protein that allosterically modulates thrombin’s enzymatic activity to favor PC activation (37). Plasminogen, a plasma-enriched zymogen, is cleaved by urokinase and tissue plasminogen activator (TPA) to generate the active enzyme plasmin, which degrades fibrin. Plasmin also cleaves PAR1 at multiple sites, which either activates or incapacitates the receptor, depending on the cleavage site (38, 39). More recently, matrix metalloproteinase-1 (MMP-1), also known as interstitial collagenase, was shown to activate PAR1 in invasive breast carcinoma, but precisely how MMP-1 acts on PAR1 to generate a functional ligand and/or cellular signaling has not been clearly established (40).
Several proteases can activate PAR2, including trypsin, FVIIa in complex with TF and FXa, neutrophil protease-2, mast cell tryptase, membrane-tethered serine protease-1 and others but not thrombin (17). The TF-FVIIa complex can activate PAR2 either directly as a binary complex or indirectly through generation of FXa. FXa may signal more efficiently in a ternary complex with TF-VIIa than it does as a monomer. Ahamed et al. showed that a TF-VIIa complex containing a distinct “cryptic” form of TF in which a specific disulfide bond is reduced fails to support coagulation (i.e., thrombin generation) but retains its ability to signal via PAR2 (41). It has not been determined, however, whether activation of PAR2 by TF-VIIa or the TF-VIIa-Xa complex promote distinct cellular responses and hence, shed doubt on the physiological relevance of these findings (42). Similar to other PARs, PAR2 is modified by N-linked glycosylation, a process that affects activation of PAR2 by tryptase but not by trypsin nor synthetic peptide agonists (43). Mutants of PAR2 in which the tethered ligand sequence SLIGRL was changed to SLAAA or SAIGRL displayed robust increases in Ca2+ mobilization when activated proteolytically by trypsin or by addition of SLIGRL synthetic peptide to unproteolyzed mutant PAR2 (44). However, when added exogenously to cells in culture, neither SLAAA nor SAIGRL synthetic peptides could elicit cellular responses comparable to those obtained with SLIGRL, the native tethered ligand sequence. These findings suggest that the cleavage event itself (which may elicit a conformational change) rather than unmasking of the ligand sequence is critical for PAR2 activation, whereas other structural determinants maybe required for activation of PAR2 by synthetic peptide agonists. The differences in signaling potential between the tethered ligand and exogenously added synthetic peptides are not clearly understood and may not be simply due to differences in the off rate of the two ligands.
Similar to other PARs, PAR4 is cleaved and activated by multiple serine proteases including thrombin, trypsin, plasmin, and cathepsin G (1). Both the kinetics of PAR4 activation and shut-off of signaling responses are slow, resulting in sustained signaling, in marked contrast to other PARs (45, 46); mechanistically how this occurs is not known. In addition, the signaling effectors and cell-type specific responses triggered by activated PAR4 remain poorly characterized. Besides thrombin, it remains to be determined whether other proteases are capable of proteolytically activating PAR3, a receptor that displays autonomous signaling (i.e., capable of signaling without input from other receptors) only in certain cell types. Clearly, many different proteases cleave and activate PARs, however, whether these proteases stabilize distinct active PAR conformations to promote coupling to specific signaling effectors and cell-type specific responses has not been thoroughly investigated. Nonetheless, new studies have revealed that activation of PAR1 by two different proteases promotes distinct cellular signaling, a phenomenon termed functional selectivity or biased agonism (6).
PAR1 Displays Biased Agonism
The finding that different ligands are capable of promoting distinct signaling responses through the activation of the same receptor has now been reported for many GPCRs (6). In many cases, differences in signaling have been observed in studies comparing synthetic ligands to natural ligands, questioning the physiological significance of such findings. Indeed, early studies suggested that distinct cellular responses could be evoked by PAR1 when activated proteolytically by its tethered ligand versus activation by untethered “free” synthetic peptide agonists (Figure 1⇑) (6, 47). More recently, McLaughlin et al. demonstrated in human endothelial cells that activation of PAR1 with thrombin favors G12/13 signaling and induction of endothelial barrier permeability rather than 2+ mobilization (48). In contrast, synthetic PAR1 Gq-dependent Ca peptide agonists SFLLRN and TFLLRNPNDK caused preferential 2+ mobilization activation favoring Gq-triggered increases in Ca rather than G12/13 signaling, but whether different endogenous proteases promote distinct signaling through the activation of PARs remained an open question.
Several recent studies have now reported that two different proteases, thrombin and activated protein C (APC), cleave and activate PAR1 but cause opposite effects on endothelial barrier permeability (Figure 2⇓) (3, 4). In general, thrombin functions as a pro-inflammatory mediator and disrupts endothelial barrier permeability through the activation of PAR1 (49). In contrast to thrombin, however, activation of PAR1 by APC elicits anti-inflammatory responses and promotes endothelial barrier stabilization (3, 4). APC is generated on the endothelial cell surface where it is poised to signal via direct activation of PAR1 (36, 50). The cyto-protective and anti-inflammatory responses induced by APC also require the co-factor function of EPCR (36). Moreover, APC reduces mortality of patients with severe sepsis (51), but the molecular mechanisms by which APC distinctly activates PAR1 signaling are not clearly understood.
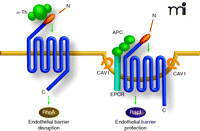
Caveolae are required for APC-mediated but not thrombin-mediated activation of PAR1 signaling. Thrombin (α-Th) activates RhoA but not Rac1 signaling and promotes endothelial barrier permeability. PAR1 is essential for thrombin –induced endothelial cellular responses but does not require caveolae for signaling. EPCR the co-factor for APC, PAR1, and G proteins associate with caveolin-1, a structural component essential for caveolae formation. In contrast to thrombin, APC activates PAR1 and induces robust Rac1 signaling but not RhoA activation, to promote endothelial barrier protection. The expression of PAR1 is required for APC cellular signaling. Moreover, APC activation of PAR1 barrier protective signaling requires compartmentalization in caveolae.
Proteolytic activation of PAR1 requires unmasking of a tethered ligand that binds intramolecularly to the body of the receptor stabilizing an active receptor conformation that triggers transmembrane signaling. Although APC has the capacity to cleave PAR1, it does so with considerably less efficiency than does thrombin (52). Thus, the extent of PAR1 activation by APC should be different than thrombin, which efficiently cleaves the receptor. Whether APC cleavage of PAR1 is the only critical determinant that facilitates PAR1 selective signaling and endothelial barrier protection is not known. If cleavage of PAR1 by APC is solely responsible for endothelial barrier–protective signaling, then quantitative, not qualitative, differences in signaling would be observed following activation of PAR1 by thrombin versus APC. To test this possibility, we recently examined the effect of thrombin and APC on the activation of RhoA and Rac1, small GTPases that differentially regulate endothelial barrier permeability (Figure 2⇑). Thrombin and APC signaling were lost in PAR1-deficient endothelial cells, indicating that PAR1 is the major effector for protease signaling in these cells (5). We also found that thrombin caused robust RhoA signaling but not Rac1 activation, whereas APC stimulated a marked increase in Rac1 activation but not RhoA signaling, consistent with the opposing functions of these proteases on endothelial barrier integrity (5). Thus, the activation of PAR1 by APC likely results in a distinct active receptor conformation that selectively couples to effectors that mediate endothelial barrier protective signaling rather than disruption.
Membrane Microdomains and Protease-Dependent Biased Agonism
How can activation of the same receptor by two different proteases elicit distinct cellular responses? The underlying mechanisms that regulate coupling of proteolytically activated PAR1 to distinct signaling effectors involve localization to caveolae, a specific type of plasma membrane microdomain (Figure 2⇑). Caveolae are lipid rafts enriched in cholesterol and caveolins and function as micro-domains that facilitate receptor-effector coupling and signal transduction. Indeed, the expression of caveolin-1 is essential for APC but not for thrombin activation of PAR1 signaling and endothelial barrier protection (5), suggesting that PAR1 localization to caveolae is critical for protease-selective signaling. The APC co-factor EPCR, PAR1, Gq, and Gi partition into lipid rafts and may exist as a preassembled complex that associates with caveolin-1 (53–55), a structural protein essential for caveolae formation in endothelial cells (56). The barrier protective signaling induced by APC is also blocked by pertussis toxin, suggesting a role for Gi/o in this process (54). Moreover, the localization of APC bound to EPCR in lipid rafts facilitates efficient PAR1 cleavage and activation, suggesting that caveolae localization stabilizes a distinct active receptor conformation that elicits barrier protective signaling (57). Taken together, these data suggest that PAR1 colocalization with EPCR and Gi/o proteins in caveolae facilitates activation by APC. In contrast, caveolin-1 is not essential for thrombin activation of PAR1 signaling (5), indicating that caveolin-1 only modulates PAR1 signaling when selectively activated by APC in endothelial cells. This finding does exclude the possibility that thrombin activates PAR1 localized to caveolae but its cellular consequences are not known. Carlie-Klusacek and Rizzo have identified a function for lipid rafts but not for caveolae in thrombin-induced changes to the cytoskeleton of endothelial cells, a cellular process controlled predominantly by G12/13 signaling (58). Moreover, caveolae are also required for TF-VIIa-mediated but not for peptide agonist–mediated activation of PAR2; but whether TF-VIIa, agonist peptide, or trypsin promote distinct cellular signaling responses was not examined (59).
The molecular determinants that specify the targeting of PAR1 and signaling components to caveolae are largely unknown but may involve post-translational modifications. Caveolin-1 is palmitoylated and together with cholesterol and sphingolipids form caveolar microdomains, which sequester other lipid-modified proteins within the plasma membrane. A large number of GPCRs appear to be modified by palmitoylation, which occurs through the covalent attachment of a C16 fatty-acid chain via a thioester linkage to cysteine residues localized within the cytoplasmic tail of the receptor. PAR1 and PAR2 have cytoplasmic cysteine residues that could serve as sites for palmitoylation, but, to our knowledge, this has not been reported. Palmitoylation of tissue factor (TF) is known to facilitate its localization to caveolae and to prevent protein kinase C–dependent phosphorylation, a process that controls tissue factor pro-coagulant activity (60). Thus, the modulation of TF with palmitoylation may facilitate localization of TF-FVIIa and Xa with PAR2 in caveolar microdomains to promote cellular signaling. Whether EPCR is similarly palmitoylated is not known. Several studies, however, do suggest that protein palmitoylation protects proteins from ubiquitination and subsequent degradation (61, 62). Both PAR1 and PAR2 are ubiquitinated (63, 64), but whether PAR ubiquitination facilitates receptor palmitoylation or targeting of proteins to caveolae has not been examined.
In addition to post-translational modifications, recent work suggests that localization of the μ-opiate receptor (MOR) to lipid rafts is regulated by its interaction with the heterotrimeric Gi2 protein [(7), see also (65)]. The localization of MOR to lipid rafts is disrupted by cholesterol depletion as well as by altering the expression of Gi2. In the absence of agonist, MOR directly associates with Gi2 via a G protein-interaction motif. Upon deletion of this motif, the receptor leaves the lipid microdomain. Etorphine promotes MOR interaction with β-arrestin, which, in turn, facilitates receptor translocation out of the lipid raft––a process that required receptor dissociation from Gi2. By contrast, stimulation of MOR with morphine induces an activate receptor conformation that preferentially binds to Gi2 and shows a low affinity for β-arrestin, consistent with minimal receptor phosphorylation and lack of internalization. Thus, membrane microdomain association, and interaction with heterotrimeric Gi2 protein regulates agonist-selective signaling by MOR. Whether PAR1 interaction with Gi2 is important for caveolae association and protease-selective signaling will be important to determine.
The expression of β-arrestin-2 is also critical for maintaining distinct assemblies of the G protein–coupled serotonin 2A receptor (5-HT2AR) with signaling effectors (66). The ablation of β-arrestin-2 expression causes selective loss of responses to the endogenous ligand serotonin, but not to a distinct synthetic ligand 2,5-dimethoxy-4-iodoamphetamine (DOI) (66). Caveolin-1 also interacts with 5-HT2AR and modulates the capacity of 5-HT2AR to couple to Gq signaling (67). Whether β-arrestin affects 5-HT2AR localization to lipid rafts was not evaluated.
Protease-Selective Mechanisms of PAR Desensitization
The ability of different ligands to promote distinct signaling responses through the activation of the same receptor suggests that unique active receptor conformations can be induced. These findings raise intriguing questions regarding the mechanisms that mediate desensitization of distinctly activated GPCRs. In the classic paradigm, activated GPCRs are initially desensitized via rapid phosphorylation by G protein–dependent kinases (GRKs) (68). Phosphorylation enhances receptor affinity for arrestin, and arrestin binding prevents receptor-G-protein interaction, thereby uncoupling the receptor from signaling. Arrestin also interacts with components of the endocytic machinery to facilitate GPCR internalization, thereby removing activated receptors from signaling effectors at the plasma membrane. Within endosomes, the receptors dissociate from their ligands, become dephosphorylated, and then return to the cell surface in a state capable of responding to ligand again. Several recent studies indicate that GPCRs activated by different ligands are desensitized through distinct mechanisms (69).
Desensitization of the μ-opioid receptor (MOR) following activation by morphine occurs predominantly through protein kinase C–dependent phosphorylation, whereas DAMGO activated MOR phosphorylation is largely dependent on GRK. Activation of the CC chemokine receptor 7 (CCR7) with the endogenous chemokine CCL19, on the other hand, induces robust phosphorylation and arrestin-dependent desensitization (70). In contrast, a different endogenous ligand, CCL21, fails to promote phosphorylation or arrestin-dependent desensitization of activated CCR7, suggesting that different ligands induce distinct desensitization of CCR7.
The desensitization of PAR1 signaling is controlled by multiple regulatory mechanisms. The first involves agonist-induced PAR1 phosphorylation and the second involves interaction with β-arrestin. Signaling by PAR1, however, appears to be more effectively regulated by β-arrestin-1 rather than by β-arrestin-2, through a process that does not require receptor phosphorylation (71, 72). Additionally, activated PAR1 internalization and lysosomal degradation are also required for termination of receptor signaling (73), whereas activated PAR1 internalization occurs independent of arrestins (72). Whether desensitization of PAR1 differs when activated proteolytically by thrombin versus synthetic peptide agonists has not been rigorously examined. We recently found that APC promotes PAR1 phosphorylation and desensitization but causes negligible receptor internalization and degradation (Figure 3⇓) (5). We have not determined whether thrombin- versus APC-activated PAR1 is differentially phosphorylated and whether distinct kinases mediate this process.
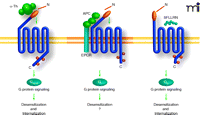
Differentially activated PAR1 is regulated by distinct mechanism. Thrombin, APC, and peptide agonists promote distinct PAR1 signaling in endothelial cells, suggesting that distinct active receptor conformations are induced. The mechanisms responsible for PAR1 desensitization following activation by distinct proteases appears be different. Activation of PAR1 by thrombin, peptide agonist, and APC promote rapid and robust receptor phosphorylation, an event critical for uncoupling PAR1 from G-protein signaling. The kinases that mediate PAR1 phosphorylation and PAR1 phosphorylation sites induced by the distinct ligands, however, have not been thoroughly examined. Moreover, thrombin and peptide agonist but not APC promotes PAR1 internalization, suggesting that the different active receptor conformations have the capacity to differentially engage the endocytic machinery. The role of arrestins or other endocytic adaptors proteins in regulating distinctly activated PAR1 internalization has not been examined.
Conclusions
PARs are activated by proteolytic cleavage, resulting in irreversible activation, unlike most GPCRs that are reversibly activated. The temporal and spatial regulation of PAR signaling is crucial for a variety of physiological responses that include vascular integrity, proper growth, and cellular migration. Activated PARs couple to multiple effectors and promote diverse cellular responses. In addition to coagulant proteases, several other proteases can cleave and activate PARs. In most cases, the signaling and cell type–specific responses elicited by different proteases remain poorly understood. That different proteases activate the same PAR but elicit distinct cellular responses (5) adds a new level of complexity to understanding PAR signaling and its functions in various tissues and cell types. Whether different proteases cleave PARs at the same or different sites to yield distinct unmasked ligands that elicit distinct cellular responses is not known. Moreover, whether the interaction of different proteases with PARs also contributions to functional selectivity remains to be determined. We will now need to further explore whether different proteases acting on the same PAR elicit distinct responses by evaluating multiple signaling effectors activated through distinct heterotrimeric G-protein subtypes as well as through non-G-protein dependent effectors. The mechanisms responsible for desensitization of PARs by different proteases have yet to be fully elucidated and are also vital to our understanding of protease signaling. Fundamental questions regarding the mechanisms by which different proteases distinctly activate PARs also remain.
The studies reported thus far, provide strong evidence that different proteases can activate the same PAR to elicit distinct signaling responses, suggesting that different drugs could be developed to target PAR signaling selectively. The ability to fine-tune PAR signaling by either activating or inhibiting specific signaling pathways could enhance therapeutic efficacies and also minimize undesirable side effects. Thus, a better understanding of how different proteases distinctly activate PAR signaling is crucial for drug development.

Acknowledgments
We thank all past and present members of the J. Trejo laboratory for comments and advice.
- © American Society for Pharmacology and Experimental Theraputics 2009
References
Unice J.K. Soh, PhD, graduated 2006 from the Department of Biological Sciences, National University of Singapore. She is currently a postdoctoral fellow in Dr. Trejo’s laboratory in the Department of Pharmacology at the University of California, San Diego and is studying the molecular basis of protease-selective activation and signaling by protease-activated receptors.

Angela Russo, MS, graduated in 2000 Magna Cum Laude from the University “Federico II” Faculty of Science, Naples, Italy. She was a graduate student in Dr. Trejo’s lab and is currently completing her dissertation studies at the University of North Carolina, Chapel Hill.

JoAnn Trejo, PhD, is an Associate Professor of Pharmacology at the University of California, San Diego. After graduating from the University of California, Davis, she completed her dissertation studies with Professor Joan Heller Brown at the University of California, San Diego. She then joined the laboratory of Professor Shaun Coughlin at the University of California, San Francisco until 1999, when she moved to the University of North Carolina, Chapel Hill as an Assistant Professor of Pharmacology. Her research program focuses on understanding the biological function and regulation of protease-activated receptors, a family of GPCRs that are uniquely activated by proteolysis. E-mail joanntrejo{at}ucsd.edu; fax 858-822-0041.
Characteristics of the Four Protease-Activated Receptors

