Designing Immunotherapies to Thwart Drug Abuse
The preferred treatment for addiction involves long-term behavioral modification programs aimed at helping patients relearn constructive behaviors, impulse control, and resistance to the craving for the drug. However, the inability to protect patients against episodic relapse to drug use is a profound challenge for most behavioral treatments. Abuse of and addiction to (+)-methamphetamine (METH) is particularly devastating and can progressively destroy individuals, families, and communities. Indeed, a study by the RAND Corporation suggests that the cost of stimulant abuse to the public was more than $23 billion in 2005 (1).
Current pharmacotherapies for managing the adverse pathological effects of METH abuse on the central nervous system, cardiovascular, and other systems are mainly supportive (2–4) and do not reverse the damage done. The rapid and sustained reductions in METH concentrations through the use of pharmacokinetic (PCKN) antagonists are a promising therapeutic strategy for clearing METH from the most vulnerable organ systems, such as the brain and heart. This medical approach uses METH-specific antibodies to sequester METH in the blood stream through high affinity binding, thereby reducing the amount and rate of drug delivery to active sites in the brain (Figure 1⇓) and other organs. In recent years, antibody medications have been investigated as a potential treatment for drug addiction and toxicity for multiple drugs, including phencyclidine, nicotine, cocaine, and METH (5–8). In this article we will highlight recent advances in this rapidly maturing field with a specific focus on antibody antagonists for METH abuse.
There are two major approaches to developing drug-specific immunotherapies: active and passive immunization. Active immunization involves conjugating a drug-like hapten to a carrier protein and using traditional immunization approaches to generate a specific immune response to the drug in the patient over time. In contrast, passive immunotherapy involves treatment with carefully selected, preformed monoclonal antibodies (mAbs) or antibody fragments against a drug of abuse. These mAbs are generated by first vaccinating a host animal and then creating mAb-secreting hybridoma cell lines, or, alternatively, by recombinant DNA methods that utilize phage, yeast, or ribosome display [see (9) for review]. These mAbs are selected for in vitro and in vivo efficacy, and if needed, converted to a human-compatible form (e.g., chimeric, humanized, or fully human immunoglobulin) (Box 1) prior to intravenous therapy in clinical scenarios. Although there are general advantages and disadvantages to both active and passive immunological approaches, not enough is known at the present time about how these antibodies will work in humans to decide which will work best. It is envisioned, however, that both treatments could have a therapeutic role in meeting the diverse medical needs of the addicted patient population.
Human-Compatible Antibodies
Antibodies are proteins and thus, can elicit unintended immune responses when administered to foreign animals. For example, monoclonal antibodies (mAbs) obtained from mouse-derived cells and hybridomas may contain amino-acid sequences not normally found in the same contiguous region of a similar human antibody. Differences in protein sequence that are recognized as foreign will likely lead to an unwanted immune response that is general to the “mouse-specific” sequences of the antibodies. Thus, a host treated with these antibodies may clear all the antibodies from the system before the salutary effect of the mAb’s METH-specific nature can occur. Fortunately, there are some techniques, beyond the scope of this Viewpoint, which can be used to minimize or eliminate the “foreignness” of antibodies raised in other organisms for use in humans.
The goal of the active immunization process is to safely stimulate high titers of antibodies with high specificity and high affinity to the target drugs. This goal is tempered by the need to avoid the creation of an adaptive immune response which, although unlikely it is still possible, could lead to the formation of antibodies that cross-react with structurally similar molecules, including endogenous compounds, over-the-counter medications, or endogenous tissues. Because drugs of abuse are too small to generate an immune response on their own, a critical step toward making an effective drug-specific vaccine is to synthesize a hap-ten that maintains the chemical and structural properties of the original drug and has an added, carefully placed chemical linker. At the distal end of the hapten linker, away from the drug moiety, is a functional group that can be easily conjugated to a larger carrier protein. In many cases, the functional group is an amine or carboxyl group for use in formation of a peptide bond with the protein. The choices for a carrier protein are diverse and can include antigenic proteins, such as keyhole limpet hemocyanin or modified bacterial toxoids that are safe for use in humans. Carrier proteins, including ovalbumin, bovine serum albumin, or many others, are used in animal models. The hapten-protein conjugate is usually admixed with an adjuvant to further heighten the immune response throughout immunization.
The proof of principle that antibodies can be used to treat drug abuse has been demonstrated in multiple preclinical and clinical scenarios dating back to 1975 when the first morphine-specific antigen was used to alter heroin self-administration by a rhesus monkey (10). Since then, the number of potential vaccines for addictive drugs has expanded greatly. For example, preclinical studies of nicotine vaccines show that the nicotine-specific antibody response leads to increases in plasma nicotine binding (11, 12) and to reductions in nicotine brain concentrations (6, 13, 14) following nicotine challenge doses. (Presumably, plasma nicotine binding was elevated and brain nicotine was reduced because the antibodies prevented nicotine from crossing into the brain.) Other studies have demonstrated that nicotine vaccines reduce nicotine-induced dopamine release and nicotine self-administration (15, 16). Rodent studies of active cocaine vaccines demonstrate similar increases in cocaine plasma concentrations and reductions in brain concentrations after cocaine challenges (7, 17, 18). In similar studies, active immunization was associated with decreases in cocaine self-administration (19) and in cocaine-induced spontaneous locomotor activity (20). The first report of a METH-specific active vaccine was by Byrnes-Blake et al. (21), in which rats were immunized with a METH-antigen vaccine in the presence of a continuous infusion of high doses of METH. The results suggested that the immune response to the METH antigen is not hindered by the presence of METH. This is an important observation because addicted patients may continue to use METH while they wait to benefit from high antibody titers that may require many (6–12) weeks of a METH-specific active vaccine immunization protocol.
Human studies of nicotine and cocaine vaccines also show that effective anti-drug titers are achieved and sustained for a period of time. At least three different nicotine vaccines have been tested in humans [see (22, 23) for further review]. In these studies, the antibody response was dose-dependent but had a large inter-subject variation (24, 25). When the immunogenic response was high, the likelihood of abstinence from nicotine increased during the time period of elevated titers (24). Similarly, in a Phase I trial of a cocaine vaccine, the patient’s cocaine-specific antibody titers following a range of antigen doses (8 to 709 μg) were shown to be dependent on vaccine dose and the number of immunological boosts (26). In a subsequent study with an even higher range of antigen doses (400–2,000 μg), there was a significantly increased incidence of cocaine-free urine samples and a higher percentage of patients reporting an attenuation of cocaine effects (27).
In a preclinical study of an METH-specific active vaccine (28), the investigators synthesized a METH-like hapten based on a prior prototype (21), and site-specifically conjugated it to a unique molecular carrier. This EP54 peptide selectively interacts with C5a receptor–bearing antigen–presenting cells, such as dendritic cells, to enhance the immune response (29). Unlike other anti-drug vaccine approaches, this study was unique because: 1) only one METH-like hapten was conjugated to the carrier peptide (unlike hapten-protein conjugates that usually have multiple hapten molecules conjugated to them) and 2) the vaccine was admixed with phosphate-buffered saline only, rather than adjuvant. Rats were vaccinated via simultaneous intraperitoneal and subcutaneous injections every seven days for five weeks, and high titers against METH were found after six weeks in response to two of the four new vaccines. It is noteworthy that the rats increased their rate of METH self-administration. Presumably the METH-specific antibodies sequestered the METH from sites of action in the brain and the rats attempted to compensate by increasing their METH intake.
MAb-based medications for treating chronic disease processes have made significant advances in the past decade with about twenty mAbs approved by the FDA and more than 150 mAbs in early or late-stage clinical trials (30). The possibility of using passive vaccines (i.e., mAbs) to treat METH addiction is in advanced stages of preclinical trials in our laboratory and nearly ready to translate to clinical trials (19, 22, 31). There are currently two forms of passive METH-specific mAbs in preclinical testing: a long-acting intact immunoglobulin G (IgG) (150 kDa) form for treating addiction and overdose, and an extremely short-acting single chain variable fragment (scFv; 27 kDa) for treatment of overdose. IgG mAb therapy for treatment of METH-like drugs (i.e., METH, amphetamine, and ecstasy) is showing promise as a treatment for METH-related addiction and overdose, and initial preclinical feasibility has been demonstrated in rats (8, 31, 32). For instance, administration of METH-specific mAbs significantly reduces METH-induced locomotor activity and shortens the duration of METH-induced peripheral effects in rats (33). As depicted in Figure 1⇓, the mAbs significantly increase serum METH concentrations while significantly decreasing METH brain concentrations (31). METH-specific mAbs also can reduce or block self-administration of METH in rats (34). Thus, preclinical studies in rats show that IgG mAbs against METH can facilitate recovery from adverse drug effects and aid in the treatment of METH abuse.
The use of passive immunotherapy has several advantages. First, mAbs with exact functional and biochemical characteristics, including high affinity and specificity, can be custom designed. Thus, an antibody with the same properties can be given to every patient in a precise dose, unlike active immunization where the immune response can vary widely between patients––a particular problem for patients that are immune compromised (e.g. HIV/AIDS patients). Second, mAbs exhibit a long half-life (6–7 days) in rats and (≤ 23 days) in humans (35). The use of a very-long-acting antagonist (2–3 weeks in duration) is a positive innovation for addicted patients who have serious problems with compliance in general, and is a missing design feature in all current small-molecule (e.g., methadone) addiction medications. Long-acting antagonists will increase the addict’s probability of success by improving patient adherence to the medical regime.
The long half-life of IgG is due partly to the ability of the constant region of the antibody to be salvaged from catabolism by the neonatal Fc receptor (called FcRn) (35, 36). Normally, IgG circulating in the vasculature is internalized into epithelial cells via pinocytosis. The endosomes are acidified and FcRn then binds to the CH2/CH3 hinge region (Figure 2⇓) of the IgG in a pH-dependent manner (pH <6.5) (37). By this process the antibody is protected from degradation and recycled to the vasculature.
If a therapeutic antibody with a shorter duration of action and greater extravascular penetration is needed, for example to treat overdose, a significantly smaller fragment lacking the constant region, such as Fab [50 kDa, half-life ranging from 0.5–21 hrs, (38)] or scFv (27 kDa, half-life ranging from minutes to hours, (39, 40)] may offer theoretical advantages (Figure 2⇓). It is also possible that a short-acting scFv could be used to rapidly clear the body of small molecule toxins. For example, Shelver et al. (41) reported desipramine-specific scFv favorably lowers serum desipramine concentrations in rats. Thus, a new and unique application for scFv is the possibility to clear drugs of abuse from the body. In a report from our group, the smaller scFv form of a METH-specific mAb was shown to effectively increase METH concentrations in the serum of rats, presumably by rapidly binding METH and re-distributing the drug from the CNS and other tissues to the blood stream. However, this redistribution of METH in vivo was not attributed to the monomeric scFv because it was eliminated or converted to multimeric forms of the scFv within a few minutes (40). Thus, although scFv has important advantages in terms of cost and ease of production, the very small molecular size (~27 kDa) of scFv monomers leads to rapid clearance (40). Nevertheless, rapid in vivo interconversion of the short-lived monomers of scFv to scFv multimers can lead to a longer duration of action (42). This PCKN property could be advantageous for treating drug overdose.
Two important properties of mAb fragments that could be improved are the PCKN profiles and the stability of multimerization of the binding sites. The small monomeric form of the scFv antibody is especially well suited for this type of customization because of the ease of design and molecular engineering into novel medications. One approach for altering the PCKN of scFv molecules is by multimerization of the scFv. This can be achieved either by linking two or more of the scFv DNA coding sequences in series, and expressing them as diabodies, triabodies, or tetrabodies (2, 3, and 4 scFv linked together, respectively), or shortening the linker to force interdomain interactions (43). These approaches have the advantage of increasing the size of the molecule, reducing the rate of clearance, and increasing the multivalency. The main drawback of this multimerization approach, however, is that despite careful design, scFv molecules tend to self-associate in unpredictable mixtures of dimers and trimers and larger molecular weight compounds (44). Thus, random conglomeration leads to problems in large-scale production and poor reproducibility of the medical properties.
Another potential strategy to customize the PCKN of scFv is to conjugate them to nanoparticles. An interesting prototype nanoparticle includes a class of molecules (1–10 nm) called dendrimers. The name dendrimer derives from the Greek dendron, meaning “tree” referring to the branched nature of the dendrimer structure that increases in density with each round of synthesis, or generation. Because of this branched structure, dendrimers possess the unique ability to carry multiple functional groups, as well as a “payload” on the interior of the molecule [see (45) for review]. They have the desirable properties of excellent monodispersity and a large number of functional termini for use in protein coupling. Researchers are beginning to explore the potential of these molecules as antibody conjugates. For example, a fifth generation dendrimer was conjugated with folate as a tumor-targeting moiety, fluorescein as a detection agent, and the cancer toxin methotrexate as a payload, all in the same molecule (46). When selectively delivered by the folate moiety to tumors in mice after biweekly injections, the methotrexate-containing dendrimer significantly lowered tumor growth rate compared to unconjugated dendrimers and the required methotrexate dose was reduced. We envision that by conjugating an METH-specific scFv to a dendrimer scaffold (Figure 2⇓), we could customize the size and thus, the pharmacokinetic profile of these newly formed “dendribodies” to create the next generation of passive immunotherapy medications. These studies are currently underway in our laboratory and could yield a new paradigm for designing immunotherapies for drug abuse.
Results from preclinical and clinical studies of active and passive vaccines against drugs of abuse show promise as a viable medical approach to treat addiction. However, antibody antagonists are not intended to be used as a standalone “magic bullet” to cure drug abuse. Similar to insulin treatment for diabetic patients, they are likely best used in combination with a long-term comprehensive medical approach. Thus, the next critical steps are to optimize the therapeutic potential and timing of active or passive immunizations and to couple these with a behavioral modification program aimed at helping patients relearn constructive behaviors, impulse control, and resistance to the craving for the drug.
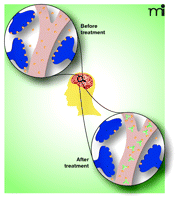
Depiction of the mechanism by which a drug-specific antibody protects the brain from adverse health effects. When drugs of abuse are self-administered, the drug (yellow circles, “Before”) rushes from the bloodstream (in gray) across the blood-brain barrier into the brain where it binds to sites of action (blue terminals) that produce euphoria. “After Treatment” with a high affinity anti-drug antibody (Y-shaped object), drug entry into the brain is restricted and rapid antibody-induced redistribution occurs which blocks or reduces the rewarding pharmacological effects. (Artwork by Misty Ward Stevens, Rachel Phillips, and Michael Owens.)
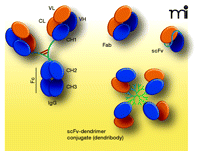
Immunoglobulin (IgG) antibody and functional fragments. The ovals represent immunoglobulin folding domains, with corresponding abbreviations: VL, variable domain light chain; VH variable domain heavy chain; CL constant domain light chain; CH, constant domain heavy chain. The polypeptide linker of the scFv is represented by a blue connecting ribbon. The dendribody is represented by a branched structure (dendrimer) with multiple scFv attached. The linker has been omitted from the scFvs of the dendribody for clarity. Figure adapted from (47) and used with kind permission of Springer Science+Business Media.
Acknowledgments
We would like to thank Dr. Nancy Rusch and the members of the Owens lab for guidance and advice during the preparation of this manuscript. Financial disclosure: S. Michael Owens serves as Chief Scientific Officer and has financial interests in InterveXion Therapeutics, LLC, a pharmaceutical biotechnology company, whose main interest is in developing new monoclonal antibodies for treatment of human diseases, including drug abuse.
- © American Society for Pharmacology and Experimental Theraputics 2009
References

Eric Peterson, PhD, is currently an Assistant Professor in the Pharmacology and Toxicology Department at UAMS. He received his doctoral degree from the University of Arkansas at Fayetteville. Subsequently, he was a NIH postdoctoral fellow in the laboratory of Dr. Michael Owens from 2002–2007, and an Instructor in the department from 2007–2009. He is the Principal Investigator of a UAMS Medical Research Endowment Grant, and an R01 grant from the National Institute for Drug Abuse that focuses on conjugating antibody fragments to dendrimers to form therapeutic dendribodies to treat drugs of abuse. He further serves as a Co-Investigator on two NIH grants with Dr. Michael Owens discovering new antibodies therapies and vaccines for the treatment of drug abuse. E-mail EPeterson{at}uams.edu; fax 502-526-4618.

S. Michael Owens, PhD, is currently Professor and the Director of the Center for Alcohol and Drug Abuse Studies, and a Professor of Pharmacology and Toxicology in the College of Medicine at the UAMS. His research interests are in translational science, antibody-based medications development, experimental therapeutics, and drug abuse. He has been continuously funded as a PI by National Institute on Drug Abuse since 1986 and was a recipient of a NIH Research Career Development Award for ten years. He has served as a grant reviewer and chairman of advisory committees for various federal research agencies, and expert panels including the NIH Small Business Innovative Research Grants program, NIDA, the NSF, the Office of Naval Technology, and the AAAS. He is a founder and Chief Scientific Officer of InterveXion Therapeutic LLC, a pharmaceutical company based-on Dr. Owens’ monoclonal antibody medications. He holds six patents and has published more than ninety publications. E-mail MOwens{at}uams.edu; fax 502-526-4618.

