|
|
Effets du tezosentan sur les symptômes et les signes cliniques des patients en insuffisance cardiaqueLes essais randomisés et comparatifs VERITAS
John J. V. McMurray, MD;
John R. Teerlink, MD;
Gadi Cotter, MD;
Robert C. Bourge, MD;
John G. F. Cleland, MD;
Guillaume Jondeau, MD;
Henry Krum, MD;
Marco Metra, MD;
Christopher M. O'Connor, MD;
John D. Parker, MD;
Guillermo Torre-Amione, MD, PhD;
Dirk J. van Veldhuisen, MD;
Jim Lewsey, PhD;
Aline Frey, PharmD;
Maurizio Rainisio, PhD;
Isaac Kobrin, MD; Pour les investigateurs VERITAS
RÉSUMÉ
| |
Contexte Les concentrations plasmatiques de l'endothéline-1,
peptide vasoconstricteur, augmentent chez les patients en insuffisance
cardiaque et des concentrations plus élevées sont
associées à un moins bon pronostic. Le tezosentan est un
antagoniste du récepteur de l'endothéline-1, à action
courte, utilisé en intraveineux, qui a des effets hémodynamiques
favorables dans l'insuffisance cardiaque.
Objectif Déterminer si le tezosentan améliore le
pronostic des patients ayant une insuffisance cardiaque aiguë.
Schéma, environnement et participants Les essais VERITAS
(Value of Endothelin Receptor Inhibition With Tezosentan in Acute Heart
Failure Studies), deux essais indépendants, randomisés, en
double aveugle, contre placebo, en groupes parallèles, ont
été menés entre avril 2003 et janvier 2005 dans ces
centres répartis en Australie, Europe, Israël et Amérique
du Nord. Les patients admis au cours des 24 heures précédents
avec une dyspnée persistante et une fréquence respiratoire de
24/minute ou plus étaient éligibles à la condition qu'ils
répondent à deux critères sur 4: (1)
élévation de la concentration plasmatique de peptide
natriurétique de type B-type ou de type N-terminal pro-B, (2) un
œdème clinique pulmonaire, (3) une congestion radiologique
pulmonaire ou un œdème, ou (4) une dysfonction systolique
ventriculaire gauche.
Traitement Perfusion de tezosentan (5 mg/heure pendant 30 minutes,
suivis de 1 mg/heure pendant 24 à 72 heures [n=730]) ou un placebo
(n=718).
Principaux critères de jugement Les critères primaires
étaient la modification de la dyspnée (mesurée aux heures
3, 6, et 24, à l'aide d'une échelle visuelle analogue
graduée de 0 à 100) sur 24 heures (comme aire sous la courbe)
dans les essais individuels et l'incidence des décès ou de
l'aggravation de l'insuffisance cardiaque au 7ème jour dans les deux
essais combinés.
Résultats Parmi les 1435 patients qui avaient reçu le
traitement assigné, 855 (60%) étaient de sexe masculin;
l'âge moyen était de 70 ans. La fraction d'éjection
ventriculaire gauche (mesurée chez 779 patients [54%]) était de
29% (DS, 11%). Les scores à l'état basal de la dyspnée
étaient similaires dans les deux groupes de traitement. Le tezosentan
n'a pas amélioré la dyspnée plus que le placebo dans l'un
ou l'autre des essais, avec une différence moyenne thérapeutique
de -12 (intervalle de confiance à 95% [IC], -105 à 81) mm/h
(P=0.80) dans le premier essai et de -25 (IC 95%, -119 à 69) mm/h
(P=0.60) dans le second. L'incidence des décès ou de
l'aggravation de l'insuffisance cardiaque au 7ème jour dans les essais
combinés a été de 26% dans chaque groupe de traitement
(rapport de cotes, 0.99; intervalle de confiance à 95%, 0.82-1.21; P
=0.95).
Conclusion Le tezosentan, antagoniste du récepteur de
l'endothéline, n'a pas amélioré les symptômes et
résultats cliniques de patients en insuffisance cardiaque
aiguë.
Trial Registration
clinicaltrials.gov
Identifiers: NCT00525707 (VERITAS-1) et NCT00524433 (VERITAS-2).
JAMA.
2007;298(17):2009-2019
L'INSUFFISANCE CARDIAQUE AIGUË est une cause fréquente
d'admission en urgence à
l'hôpital.1-3
Le symptôme principal, la dyspnée, est probablement due
à une augmentation de la pression capillaire pulmonaire (PCWP), souvent
associée à une diminution du volume d'éjection et de
l'index cardiaque ainsi qu'à une augmentation des résistances
systémiques
vasculaires.1-3
Les objectifs immédiats du traitement sont de soulager la
dyspnée et d'améliorer et de stabiliser l'état
hémodynamique et clinique du
patient.1-5
Les autres objectifs sont la prévention du décès et des
réadmissions, lesquels surviennent
fréquemment.1-5
Actuellement, les patients en insuffisance cardiaque aiguë sont
traités avec des médicaments ayant une action diurétique,
vasodilatatrice ou inotropique (ou certaines combinaisons de ces actions),
bien que la base des preuves pour l'un ou l'autre des traitements dans
l'insuffisance cardiaque aiguë soit
faible.1-5
Les endothélines sont des peptides ayant une action
vasoconstrictrice puissante ainsi que d'autres effets qui peuvent être
délétères dans l'insuffisance
cardiaque.6-8
L'élévation des concentrations d'endothéline plasmatique
est un facteur prédictif de pronostic péjoratif chez les
patients ayant une insuffisance
cardiaque.6-8
Le tezosentan est un antagoniste des récepteurs de type A et de type B,
à action courte, développé pour une utilisation
intraveineuse.9-13
C'est un vasodilatateur puissant qui réduit les résistances
vasculaires systémiques et la PCWP et augmenté le débit
cardiaque de manière
dose-dépendante.9-13
Les études VERITAS (Value of Endothelin Receptor Inhibition With
Tezosentan in Acute Heart Failure Studies) ont été
conçues pour évaluer l'hypothèse que le tezosentan aurait
un effet favourable sur les symptômes et les résultats cliniques
chez des patients en insuffisance cardiaque
aiguë.14
METHODES
Le programme VERITAS comprenait deux études indépendantes,
identiques et concurrentes (VERITAS-1 et VERITAS-2) menées entre avril
2003 et janvier 2005, dont le schéma a été décrit
en
détail.14
L'étude a été approuvée par le comité
d'éthique/bureau indépendant de revue de chaque hôpital
participant.
Le comité scientifique, un groupe de conseillers, et le sponsor ont
conçu les essais et se sont périodiquement rencontrés
pour évaluer les progrès de l'étude, résoudre les
questions opérationnelles et, à la fin de l'étude,
interpréter les résultats. Le bureau indépendant du suivi
des données et de la tolérance
(DSMB)14
s'est réuni périodiquement pour revoir les données de
l'étude et, après une analyse intermédiaire, pour revoir
les données d'efficacité. Le DSMB avait la possibilité de
recommander de mettre fin précocement au programme de l'étude
(voir ci-dessous) si des questions majeures survenaient concernant la
tolérance du tezosentan. L'analyse des données pour le DSMB
était effectuée par un groupe de statisticiens
indépendants.
Schéma de l'étude
VERITAS-1 et VERITAS-2 étaient des études randomisées,
en double aveugle, contre placebo et en groupes parallèles
(FIGURE 1). Les patients
étaient éligibles s'ils avaient 18 ans ou plus et
répondaient aux critères d'inclusion et d'exclusion. Le
critère majeur d'inclusion était l'admission à
l'hôpital avec une insuffisance cardiaque aiguë au cours des 24
heures précédentes et la persistance d'une dyspnée au
repos. Le rapport du patient sur la dyspnée devait être
appuyé par une fréquence respiratoire d'au moins 24/minute. Le
diagnostic clinique de l'investigateur d'une insuffisance cardiaque devait
être appuyé par au moins 2 des 4 critères suivants: (1)
élévation de la concentration de peptide natriurétique de
type B (BNP) ou N-terminal pro-BNP, (2) oedème pulmonaire à
l'examen physique, (3) congestion radiologique pulmonaire ou oedème, ou
(4) dysfonction ventriculaire systolique gauche (fraction d'éjection
ventriculaire gauche [FEVG] <40% ou indice cinétique pariétal
 1.2). Si le patient avait un cathéter dans l'artère
pulmonaire, l'index cardiaque (mesuré en l/min par m2) devait
être 2.5 et la PCWP 20 mm Hg ou plus. 1.2). Si le patient avait un cathéter dans l'artère
pulmonaire, l'index cardiaque (mesuré en l/min par m2) devait
être 2.5 et la PCWP 20 mm Hg ou plus.
|
|
|
|
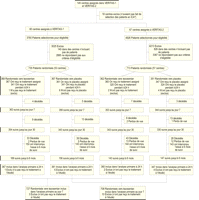
Figure 1.. Disposition des patients dans VERITAS-1 et VERITAS-2
VERITAS correspond à "Value of Endothelin Receptor
Inhibition With Tezosentan in Acute Heart Failure Studies"
*ICA correspond à insuffisance cardiaque aiguë
|
|
|
Le patient devait aussi avoir reçu au moins une dose de
diurétique intraveineux 24 heures ou moins et 2 heures ou plus avant le
début du traitement à l'étude (ou une perfusion de
diurétique à un taux constant 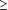 2 heures). Si les autres
traitements intraveineux avaient été initiés avant le
début du traitement à l'étude, leur dose devait
être stable pendant deux heures ou plus pour les vasodilatateurs, les
sympathomimétiques, ou les sensibilisateurs calciques ou pendant 4
heures pour les inhibiteurs de la phosphodiestérase et le
nésiritide. 2 heures). Si les autres
traitements intraveineux avaient été initiés avant le
début du traitement à l'étude, leur dose devait
être stable pendant deux heures ou plus pour les vasodilatateurs, les
sympathomimétiques, ou les sensibilisateurs calciques ou pendant 4
heures pour les inhibiteurs de la phosphodiestérase et le
nésiritide.
Les critères d'exclusion avaient été décrits
dans le detail et incluaient le choc cardiogénique dans les 48 heures,
un IDM avec élévation du segment ST, une ischémie
évolutive ou l'administration d'un agent throm-bolytique, une
hypotension (pression artérielle systolique <100 mg Hg chez les
patients ne recevant pas de vasodilatateur ou <120 mm Hg chez ceux recevant
un vasodilatateur), une anémie (concentration d'hémoglobine
<10 g/dl, hématocrite<30%), ou dysfonction rénale
(créatininémie > 2.5 mg/dl [pour convertir en µmol/l,
multiplier par
88.4]).14
Les informations sur la race/ethnie étaient recueillies pour chaque
patient au moyen d'un check-list par l'investigateur.
Traitement à l'étude
Après le recueil du consentement éclairé écrit,
les patients étaient randomisés 24 heures ou moins après
admission en vue de recevoir une perfusion soit de placebo soit de tezosentan,
5 mg/h pendant 30 minutes, suivie par 1mg/h pendant au moins 24 (et
jusqu'à 72) heures. Les patients du groupe placebo recevaient un volume
identique de perfusion de sérum salé (104 ml pendant les 24
premières heures). Les patients et les investigateurs de l'étude
travaillaient en aveugle par rapport à l'assignation aléatoire
de l'étude.
Critères primaires de jugement
Le critère primaire de jugement pour chaque patient de
l'étude VERITAS était la modification de la dyspnée par
rapport à l'état basal durant les 24 premières heures de
traitement, mesurée aux heures 3, 6, et 24 sous la forme de l'aire sous
la courbe. La dyspnée était évaluée par chaque
patient à l'aide d'une échelle visuelle analogique (VAS)
consistant de lignes verticales de 10 cm avec la phrase « je ne suis pas
essoufflé du tout » en bas de l'échelle et « je suis
plus essoufflé que je ne l'ai jamais été » en haut
de l'échelle. La VAS était cotée de 0 à 100, mais
le patient ne connaissait pas la valeur numérique de sa
réponse.
Le critère d'efficacité primaire des études combines
était l'incidence des décès ou de l'aggravation de
l'insuffisance cardiaque au 7ème jour. L'aggravation de l'insuffisance
cardiaque pouvait survenir au cours de l'admission initiale ou index ou
après la sortie. Une aggravation de l'insuffisance cardiaque au cours
de l'admission initiale était définie soit par le
développement d'un oedème pulmonaire, un choc
cardiogénique ou d'autres signes d'aggravation de l'insuffisance
cardiaque ou comme une absence d'amélioration de l'insuffisance
cardiaque du patient sous traitement (échec du traitement). Les deux
définitions demandaient au moins un élément parmi les
éléments suivants: (1) initiation d'une nouvelle thérapie
intraveineuse, (2) ré-institution de la thérapie intraveineuse
précédente, (3) augmentation du traitement intraveineux en cours
pour insuffisance cardiaque, (4) mise en place d'une assistance
mécanique circulatoire (pompe de contre-pulsation intra-aortique) ou
respiratoire (y compris avec pression continue positive respiratoire), ou (5)
utilisation d'une ultrafiltration, d'une hémofiltration, ou d'une
hémodialyse. L'aggravation de l'insuffisance cardiaque survenant
après l'admis- sion index était définie soit par une
visite non programmée aux Urgences en raison d'une aggravation de
l'insuffisance cardiaque (oedème pulmonaire, choc cardiogénique,
ou autre signe d'aggravation) ou pour admission non programme en raison d'une
aggravation de l'insuffisance cardiaque. Les deux définitions
demandaient au moins un des éléments suivants: (1)
administration d'un traitement intraveineux pour insuffisance cardiaque,
incluant diurétiques, vasodilatateurs, ou agents inotropes positifs;
(2) mise en place d'une assistance mécanique circulatoire ou
respiratoire; ou (3) utilisation d'une ultrafiltration, hémofiltration,
ou hémodialyse.
Principaux critères secondaires
Les objectifs secondaires étaient de déterminer si le
tezosentan diminuait l'incidence des décès ou des
événements cardio-vasculaires majeurs au 30ème jour,
améliorait les constantes hémodynamiques au cours des 24
premières heures (dans le sous-groupe de patients ayant un
cathéter pulmonaire en place pour indications cliniques), diminuait la
durée du séjour hospitalier initial et le nombre de jours
à l'hôpital jusqu'au 30ème jour et influençait la
mortalité à 6
mois.14
Analyses statistiques
Le critère principal des études individuelles, l'aire sous la
courbe montrant la modification du score de dyspnée au cours des 24
premières heures de traitement, était analysé comme une
variable normalement distribuée (avec une déviation standard
attendue de 600 mm/h). L'hypothèse nulle était testée au
moyen d'un test t contre une alternative d'une différence moyenne d'au
moins 150 mm/h pour le groupe actif par rapport au groupe placebo. Le
critère primaire des deux études combinées, l'incidence
des décès ou l'aggravation de l'insuffisance cardiaque, a
été analyse comme une variable binomiale. L'hypothèse
nulle a été évaluée à l'aide d'un test
exact de Fisher contre l'alternative d'une réduction du risque de 25%
lorsque le groupe actif était comparé au placebo (avec un taux
attendu d'événements de 35%%).
L'erreur globale de type I de 0.05 dans le programme VERITAS était
divisée, 0.0008 étant attribué à l'analyse de
chaque étude individuelle (0.04 chaque) et 0.0492 (ou 0.0092 en
utilisant une erreur globale de type I plus restrictive de 0.01) aux
études combinées. L'intention était de soumettre le
programme VERITAS pour enregistrement auprès de la FDA (US Food and
Drug Administration) pour approbation commerciale si le critère des
décès ou de l'aggravation de l'insuffisance cardiaque atteignait
un niveau de signification inférieur à 0.01 dans l'analyse des
études combinées ou si le critère dyspnée
était significatif à un niveau inférieur à 0.04
dans l'analyse de chaque étude individuelle (VERITAS-1 et VERITAS-2) ou
si le critère des décès ou de l'aggravation de
l'insuffisance cardiaque était significatif pour une valeur  globale de 0.05. Sur la base des conditions ci-dessus et avec une puissance
90% pour chacune des analyses des critères primaires, la taille de
l'échantillon requise a été estimée être de
1760 patients (440 dans chaque groupe des deux études). L'analyse
était en intention de traiter. Des analyses intermédiaires des
critères primaires étaient programmées pour être
réalisées après la randomisation de 50% et 75% des
patients. Le DSMB pouvait recommander l'interruption de l'étude (pour
futilité de l'hypothèse) s'il devenait évident lors de
l'analyse intermédiaire qu'aucun des objectifs du programme ne pourrait
être atteint. L'interruption serait conseillée si la puissance
conditionnelle (calculée à l'aide de la méthode de
raccourcissement stochastique) tombait au-dessous de 10% à la fois pour
les décès ou pour l'aggravation de l'insuffisance cardiaque au
7ème jour (pour un = 0.05) et de la dyspnée (pour un
= 0.04 pour chaque
étude).14
Ces conditions ont été remplies lors de la deuxième
analyse intermédiaire, et la randomisation a été
interrompue le 6 novembre 2004. Les analyses ont été
effectuées à l'aide du logiciel SAS version 8.1 (SAS Institute
Inc, Cary, Caroline du Nord).
globale de 0.05. Sur la base des conditions ci-dessus et avec une puissance
90% pour chacune des analyses des critères primaires, la taille de
l'échantillon requise a été estimée être de
1760 patients (440 dans chaque groupe des deux études). L'analyse
était en intention de traiter. Des analyses intermédiaires des
critères primaires étaient programmées pour être
réalisées après la randomisation de 50% et 75% des
patients. Le DSMB pouvait recommander l'interruption de l'étude (pour
futilité de l'hypothèse) s'il devenait évident lors de
l'analyse intermédiaire qu'aucun des objectifs du programme ne pourrait
être atteint. L'interruption serait conseillée si la puissance
conditionnelle (calculée à l'aide de la méthode de
raccourcissement stochastique) tombait au-dessous de 10% à la fois pour
les décès ou pour l'aggravation de l'insuffisance cardiaque au
7ème jour (pour un = 0.05) et de la dyspnée (pour un
= 0.04 pour chaque
étude).14
Ces conditions ont été remplies lors de la deuxième
analyse intermédiaire, et la randomisation a été
interrompue le 6 novembre 2004. Les analyses ont été
effectuées à l'aide du logiciel SAS version 8.1 (SAS Institute
Inc, Cary, Caroline du Nord).
|
|
|
|
Figure 2.. Modification de la dyspnée dans VERITAS-1 et VERITAS-2
Les modifications de la dyspnée montrées sont les aires sous
la courbe. VERITAS correspond à "Value of Endothelin Receptor
Inhibition With Tezosentan in Acute Heart Failure Studies". Les barres
d'erreur correspondent à l'intervalle de confiance à 95%.
|
|
|
RESULTATS
Entre le 11 avril 2003 et le 20 janvier 2005, 1448 patients ont
été randomisés et évalués dans 110 centres
en Australie, Europe, Israël, et Amérique du Nord: 735 patients
dans 53 centres pour l'étude VERITAS-1 et 713 dans 57 centre pour
l'étude VERITAS-2. En raison de l'interruption prématurée
des essais pour futilité, seulement 82% du nombre prévu de
patients (1760) a été inclus. La distribution des patients dans
les essais est résumée dans la
Figure 1.
Caractéristiques initiales
Les groupes de traitement étaient bien équilibrés pour
toutes les caractéristiques. Des 1435 patients ayant reçu le
traitement tel qu'assigné, 855 (60%) étaient de sexe masculin;
l'âge moyen était de 70 ans. Une insuffisance cardiaque
était présente avant l'admission chez 1045 patients (73%), et
750 (52%) avaient des antécédents d'infarctus du myocarde; 1139
(79%) avaient des antécédents d'hypertension artérielle,
et la pression artérielle moyenne systolique à l'état
basal était de 132 mm Hg (TABLEAU
1). La fraction d'éjection ventriculaire a
été mesurée chez 779 patients (54%); la FEVG moyenne (DS)
était de 29% (11%). Les investigateurs ont réalisé un
suivi hémodynamique invasif chez 84 patients (6%). L'index cardiaque
moyen (intervalle de confiance 95% [IC]) était de 2.06 (1.97 à
2.18); la PCWP, 26 (25 à 27) mm Hg; et les résistances
vasculaires systémiques, 1777 (1630 à 1926)
dyne•s/cm5.
|
|
|
|
Tableau 1.. Caractéristiques cliniques des patients dans VERITAS-1 et
VERITAS-2
Abréviations: IEC, inhibiteur de l'enzyme de conversion; BNP,
peptide natriurétique de type B; FEVG, fraction d'éjection
ventriculaire gauche; NT pro-BNP, N-terminal pro-BNP; ULN, limite
supérieure de la normale; VERITAS, "Value of Endothelin Receptor
Inhibition With Tezosentan in Acute Heart Failure Studies".
Facteurs de conversion IS: Pour convertir les valeurs de la natrémie
en mmol/l, multiplier par 1.0; les valeurs de l'azotémie en mmol/L, par
0.357; les valeurs de la créatininémie en µmol/L, par 88.4;
les valeurs de l'hémoglobine en g/L, par 10.0.
aPatients prenant un diurétique de l'anse au moment de la
randomisation (43% recevaient un diurétique de l'anse à ce
moment); per protocole, 99% des patients avaient reçu un
diurétique de l'anse par voie intraveineuse 24 and 2 heures
avant l'initiation du traitement à l'étude.
bAdministré dans les 24 heures avant l'initiation du
traitement à l'étude. Furosémide (20 mg) =
bumétanide (1 mg), acide éthacrynique (25 mg), torasémide
(10 mg), et pirétanide (6 mg).
|
|
|
Critères d'inclusion dans l'étude
La majorité des 1435 patients ont été éligibles
dans l'étude sur la foi de signes cliniques (1284 [90%]) et
radiologiques (1185 [83%]) de congestion pulmonaire ou d'oedème. Peu
ont été inclus sur la base d'une dysfonction ventriculaire
systolique documentée (677 [47%]) ou sur une élévation de
la concentration du BNP (262 [18%]) ou du N-terminal pro-BNP (32 [2%]).
|
|
|
|
Figure 3.. Modification de la dyspnée sur 24 heures dans les sous-groupes
clés de VERITAS-1 et VERITAS-2
Les modifications de la dyspnée montrées sont les aires sous
la courbe. VERITAS correspond à "Value of Endothelin Receptor
Inhibition With Tezosentan in Acute Heart Failure Studies". IC
correspond à intervalle de confiance; VAS, Echelle visuelle analogique.
Les barres d'erreur correspondent à l'intervalle de confiance à
95%.
|
|
|
Traitement à l'état de base
Comme programmé, tous les patients ont reçu un
diurétique intraveineux. Au moment de la randomisation, un inhibiteur
de l'enzyme de conversion de l'angiotensine ou un antagoniste du
récepteur de l'angiotensine a été utilisé chez 909
patients (63%), un bêtabloquant chez 671 (47%), et de la spironolactone
chez 265 (19%). Très peu de patients ont reçu d'autres agents
vaso-actifs intraveineux à l'état de base ou au cours de la
perfusion du médicament à l'étude: 221 (15%) ont
reçu des dérivés nitrés à l'état de
base (20% au cours de la perfusion du traitement à l'étude), 26
(1.8%) ont reçu de la dobutamine (3.3%), 7 (0.5%) du nitroprusside
(1.1%), 21 (1.5%) du nésiritide (2.9%), 29 (2.0%) de la dopamine
(4.5%), et 2 (0.1%) de la milrinone (0.6%).
Administration du traitement à l'étude
Le traitement à l'étude a été reçu selon
l'assignation par 727 des 730 (99.6%) patients assignés pour recevoir
tezosentan et 708 des 718 patients (98.6%) assignés pour recevoir le
placebo (Figure 1). Le temps
médian entre l'admission et le début du traitement à
l'étude a été de 11 heures. Au moins 24 heures de
traitement ont été reçus par 661 des 727 (91%) patients
sous tezosentan et 665 des 708 patients (94%) sous placebo.
Modification de la dyspnée
Le score moyen (IC 95%) de la VAS pour la dyspnée à
l'état basal était de 66.5 (64.2 à 68.8) dans le groupe
tezosentan et 63.7 (61.5 à 65.9) dans le groupe placebo à
l'état basal dans VERITAS-1; les scores correspondants dans VERITAS-2
étaient de 60.3 (57.7 à 62.9) et de 59.4 (56.7 à 62.0),
respectivement, un score élevé indiquant une dyspnée plus
importante.
Le niveau de dyspnée a diminué rapidement dans les deux
groupes de traitement après la randomisation
(FIGURE 2). Le critère
primaire de l'aire sous la courbe pour la modification de la dyspnée
par rapport à l'état basal était similaire dans chaque
groupe de traitement à la fois dans VERITAS-1 et VERITAS-2. Dans
VERITAS-1, la modification moyenne (IC 95%) était de -562 (-628
à -497) mm•h dans le groupe tezosentan et de -550 (-617 à
-483) dans le groupe placebo (différence entre les traitements, -12
[-105 à 81]mm•h; P=0.80). Dans VERITAS-2, la modification
correspondante dans le groupe tezosentan était de -367 (-432 à
-302) mm•h et dans le groupe placebo de -342 (-410 à -274)
(différence entre traitements, -25 [-119 à 69] mm•h;
P=0.60).
Il n'y a eu aucune différence entre le tezosentan et le placebo dans
les sous-groupes clés non définis
(FIGURE 3). La réponse
des patients ayant des scores VAS à l'état basal pour la
dyspnée supérieurs et inférieurs à la
médiane (65 mm) a été similaire. De même, une
analyse prédéfinie sur la modification de la fréquence
respiratoire (analyse de l'aire sous la courbe) ne montrait aucune
différence entre le tezosentan et le placebo; dans VERITAS-1 et
VERITAS-2 combinées, la modification moyenne (IC 95%) dans le groupe
tezosentan était de -32 (-50 à -15) respirations/min•h et
dans le groupe placebo de -26 (-43 à -9) (différence entre les
traitements, -6.4 [-31 à 18]; P=0.61).
La modification moyenne (IC 95%) de l'aire sous la courbe pour
l'évolution de la dyspnée n'a pas différé entre
les groupes de traitement pour le sous-groupe des 84 patients suivis
hémodynamiquement (différence de traitement, -5 [-328 à
318]; P=0.97), et la différence thérapeutique n'a pas
différé significativement entre les patients qui étaient
et qui n'étaient pas suivis hémodynamiquement (P=0.89 pour
l'interaction).
Décès ou aggravation de l'insuffisance cardiaque durant la période de 7 jours
L'incidence du critère primaire des décès ou
aggravation de l'insuffisance cardiaque au cours des 7 premiers jours suivant
la randomisation dans VERITAS-1 et VERITAS-2 combinés est
montrée dans le TABLEAU
2. Dans les groupes de traitement, 26% des patients ont
présenté ce critère composite (rapport de cotes, 0.99; IC
95%, 0.82 à 1.21; P=0.95). L'effet du tezosentan par rapport au placebo
a été similaire dans tous les sous-groupes non
prédéfinis y compris ceux définis par le sexe,
l'âge, l'étiologie, la FEVG, la comorbidité, la pression
artérielle initiale, la fonction rénale et le traitement
concomitant.
|
|
|
|
Tableau 2.. Critères primaires (Décès ou aggravation de
l'insuffisance cardiaque) et secondaires (Décès ou
événement cardio-vasculaire majeure) jusqu'au 7ème jour
et à jour 30.
aComparaison entre les groupes de traitement (Test exact de
Fisher): P = 0.95 au jour 7 et P = 0.61 au jour 30.
bClassés par gravité; chaque patient comptait pour
un (l'événement le plus grave); analyses séparées
pour les jours 7 et 30, un patient en choc cardiogénique au jour 6 qui
décédait le jour 8 était classé comme ayant un
choc cardiogénique à l'analyse du jour 7 et comme
décès à l'analyse du jour 30. Les patients qui avaient
une transplantation cardiaque ou étaient perdus de vue étaient
comptabilisés comme échecs de traitement.
cComparaison entre groupe de traitement (Test exact de Fisher):
P = 0.95 au jour 7 et P = 0.66 au jour 30.
dChaque patient pouvait avoir plus d'un
événement.
|
|
|
Critères secondaires et exploratoires
Le critère secondaire sur l'incidence des décès ou
aggravation de l'insuffisance cardiaque à 30 jours a été
de 32% dans le groupe tezosentan et de 33% dans le groupe placebo
(Tableau 2).
L'incidence du critère composite majeur à 7 jours et 30 jours
est résumée dans le Tableau
2.
Le nombre moyen (DS) de jours passés à l'hôpital au
cours de l'admission index après la randomisation a été
de 9 (7) dans les deux groupes de traitement et de la randomisatoin au jour
30, de 11 (8) dans chaque groupe de traitement. Le nombre de
décès à 6 mois a été de 104 (14.3%) dans le
groupe tezosentan et de 101 (14.3%) dans le groupe placebo (risque relatif,
1.01; IC 95%, 0.77 à 1.33).
Modifications des mesures hémodynamiques
Les modifications des mesures clés hémodynamiques aux heures
3, 6, et 24 à partir du début de traitement à
l'étude constituaient les critères secondaires et sont
résumées dans le TABLEAU
3. Des diminutions plus importantes de la PCWP, de la pression
auriculaire droite, des résistances vasculaires pulmonaires et
systémiques et une tendance à une augmentation plus importante
de l'index cardiaque ont été observées dans le groupe
tezosentan.
|
|
|
|
Tableau 3.. Modifications hémodynamiques par rapport à l'état
basal à 3, 6, et 24 heures chez les patients ayant eu un
cathéter de l'artère pulmonaire
Abréviations: IC, Intervalle de confiance; mPAP, pression moyenne de
l'artère pulmonaire; PCWP, pression capillaire pulmonaire; PVR,
résistance vasculaires pulmonaires; RAP, pression oreillette droite;
SVR, résistances vasculaires systémiques. Toutes les valeurs
sont des moyennes (erreur standard ou moyenne).
aDu test U de Mann-Whitney.
bCalculé comme l/min par m2
|
|
|
A 24 heures, la diminution moyenne (DS) de la pression artérielle
systolique était de 14.6 (18.3) mm Hg dans le groupe tezosentan et de
-8.5 (21.1) mm Hg dans le groupe placebo, une différence
statistiquement significative de -6.1 (IC 95%, -8.2 à -4.1) mm Hg
(P<0.0001). La diminution moyenne (DS) de la pression artérielle
diastolique a été de -9.7 (13.7) mm Hg dans le groupe tezosentan
et de -4.4 (14.3) mm Hg dans le groupe placebo, une différence
statistique significative de -5.3 (IC 95%, -6.7 à -3.8) mm Hg
(P<0.0001).
Sécurité et effets indésirables
Parmi les 727 patients traités par tezosentan et les 708 patients
traités par placebo, 103 (14.2%) et 68 (9.6%), respectivement, ont
stoppé le médicament à l'étude en raison d'un
effet indésirable (P=0.008). Cette différence a
été principalement due à une hypotension
(entraînant l'interruption du traitement chez 60 patients [8.3%] dans le
groupe tezosentan et chez 33 [4.7%] dans le groupe placebo, P=0.003); ceci a
été l'effet indésirable le plus fréquent
associé à l'interruption de traitement. Globalement, une
hypotension persistant jusqu'à 5 jours après l'initiation du
medicament à l'étude a été rapporté comme
effet indésirable chez 165 patients du groupe tezosentan (22.7%) et
chez 103 du groupe placebo (14.5%) (P<0.001). A 72 heures, l'augmentation
moyenne de l'azotémie du groupe tezosentan était de 1.7 (5.0)
mmol/l; dans le groupe placebo, elle était de 1.5 (6.2) mmol/l
(différence moyenne, 0.2; IC 95%, -0.4 à 0.8; P=0.58). Pour le
même temps de mesure, l'augmentation moyenne de la
créatininémie a été de 0.09 (0.35) mg/dl dans le
groupe tezosentan et de 0.07 (0.32) mg/dl dans le groupe placebo
(différence moyenne, 0.02; IC 95%, -0.02 à 0.05; P=0.39).
COMMENTAIRE
Dans VERITAS, une faible dose de tezosentan n'a pas réussi à
améliorer la dyspnée de patients examinés
précocement après une admission non programmée dans un
hôpital pour insuffisance cardiaque aiguë, et n'a pas réussi
à améliorer le risque ultérieur de décès ou
d'événements non mortels cardio-vasculaires. La dose de
tezosentan utilisée dans VERITAS a été choisie sur la
base d'une étude d'ajustement de dose contre placebo qui montrait que
cette dose pouvait diminuer la PCWP et les résistances vasculaires
systémiques de même que les concentrations plasmatiques de
BNP.13 Ces
actions hémodynamiques ont été confirmées dans
VERITAS par la réduction de la pression artérielle et, dans un
sous-groupe de patients, par celle de la PCWP.
Pourquoi le tezosentan n'a pas réussi à améliorer la
dyspnée dans VERITAS, en dépit des preuves d'amélioration
hémodynamique? La réduction de la PCWP dans un sous-groupe de
patients VERITAS porteurs d'un cathéter de l'artère pulmonaire a
été de la même importance que celle produite par le
tezosentan dans les études
antérieures.9,12,13
Ce qui est plus important, c'est qu'elle a été aussi similaire
au changement de la PCWP observe dans les essais sur le levosimendan et le
nésiritide, dans lesquels ces deux traitements diminuaient la
dyspnée chez des patients suivis de façon
invasive.15,16
Il est toutefois possible que l'effet hémodynamique de la dose de
tezosentan utilisée ait été présent chez les
patients non suivis de VERITAS. Le tezosentan a aussi diminué la
dyspnée et la PCWP plus que le placebo chez les patients suivis
invasivement dans l'étude RITZ-2 (Randomized Intravenous Tezosentan 2)
(n=292). L'inclusion nécessitait un index cardiaque faible ( 2.5)
et une augmentation de la PCWP ( 15mmHg). Bien qu'une dose plus importante
de tezosentan (50 ou 100 mg/h) ait été perfusée dans
RITZ-2 que dans VERITAS (1 mg/h), l'effet du tezosentan sur la PCWP dans
RITZ-2 (réduction moyenne corrigé contre placebo, 4.1 mm Hg) a
été similaire à celui du sous-groupe de patients ayant eu
un suivi invasif hémodynamique dans VERITAS (réduction moyenne
corrigé contre placebo, 3.3 mm
Hg).17 Une
explication propose pour la différence apparente entre VERITAS et ces
essais antérieurs est que la connaissance des modifications
hémodynamiques a d'une certaine façon influence
l'évaluation de la
dyspnée.18
Dans un essai sur le nésiritide qui ne s'accompagnait pas de suivi
hémodynamique invasif, aucune des doses de nésiritide n'a
amélioré la dyspnée plus que le traitement
habituel.16
De plus, dans l'essai RITZ-1, dans lequel les patients (n=669) n'avaient pas
eu de suivi hémodynamique invasif, le traitement par tezosentan (50
mg/h) n'a pas amélioré la
dyspnée.19
De même dans un large essai contre placebo sur le levosimendan qui ne
comportait pas de suivi hémodynamique, il n'y a pas eu
d'amélioration apparente de la
dyspnée.20
Dans un autre essai, l'étude VMAC (Vasodilation in the Management of
Acute Congestive Heart Failure), le nésiritide (mais pas le
glycéryl trinitrate) a amélioré la dyspnée
à 3 heures, mais une revue de la FDA (Food and Drug Administration) a
conclu que cette différence était due à un sous-groupe de
patients suivis
hémodynamiquement.21,22
Les résultats de trois autres essais, semblent toutefois
réfuter l'hypothèse que la connaissance des modifications
hémodynamiques puisse influencer l'évaluation de la
dyspnée, bien qu'une comparaison avec et sans suivi soit seulement
disponible pour le nésiritide, le tezosentan, et le levosimendan. Dans
l'essai REVIVE (Randomized Multicenter Evaluation of Intravenous Levosimendan
Efficacy), il a été rapporté que le levosimendan
améliorait la dyspnée comparé au placebo, mais d'autres
détails sur le pourcentage de patients suivis invasivement, et la
modification de la dyspnée chez ces patients comparés aux
patients non suivis, n'ont pas encore été
présentés.23
De plus, 2 essais récents à grande échelle, contre
placebo ont montré que l'antagoniste de l'arginine vasopressine, le
tolvaptan, améliorait la dyspnée rapportée par le
patient, apparemment sans que l'investigateur ait connaissance de
l'hémodynamique.24
Une autre explication est que l'effet potentiellement
bénéfique sur l'essoufflement de la réduction de la PCWP
par le tezosentan peut avoir été déclenché par une
autre action négative du blocage de l'endothéline, par exemple
l'induction d'un shunt pulmonaire artério-veineux conduisant à
une désaturation.
Nous ne pensons pas que d'autres facteurs expliquent la différence
des observations entre VERITAS et les essais "positifs" sur le
nésiritide et le levosimendan. Les patients dans VERITAS étaient
en général similaires bien qu'un tout petit peu plus
âgés, et un pourcentage plus élevé avait une FEVG
préservée. Toutefois, l'effet du tezosentan était
similaire dans tous les sous-groupes, y compris ceux définis par
l'âge et le FEVG. Le sous-groupe qui a eu un suivi hémodynamique
dans VERITAS avait la même PCWP à l'état basal que les
patients d'ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary
Artery Catheterization
Effectiveness).25
Il a été montré que la VAS était une mesure
plus sensible et plus reproductible de la modification de la dyspnée
que les échelles de Likert et Borg, bien que dans un contexte autre que
celui de l'insuffisance cardiaque
aiguë.26
Il est préoccupant, toutefois, que l'évaluation du
symptôme clé dans l'insuffisance cardiaque aiguë, la
dyspnée, ait été si mal étudié. Ceci
constitue une limite majeure méthodologique des essais dans
l'insuffisance cardiaque aiguë et l'importante amélioration de la
dyspnée dans le groupe placebo (en réponse aux soins habituels)
rend difficile de détecter tout autre effet des nouveaux traitements
par les outils communément employés, la VAS et l'échelle
de Likert. Dans les études récentes tolvaptan, un score
rapporté par le patient était utilise, et ceci a permis de
détecter une différence entre le traitement actif et le
placebo.24
Comprendre si l'efficacité diffère avec ces différents
instruments dans l'évaluation de l'essoufflement sera important pour la
conduite des futurs essais dans ce domaine.
Le tezosentan n'a eu aussi aucun effet sur le critère composite
mortalité/morbidité (ou sur l'un quelconque des critères
secondaires), bien que même avec ce critère, nous ayons eu une
puissance de 80% pour détecter une réduction du risque relatif
de 25%. Bien que décevantes et contraires aux attentes basées
sur le rôle potentiel physiopathologique de l'endothéline-1 dans
l'insuffisance cardiaque, nos observations sont compatibles avec les essais
antérieurs dans l'insuffisance cardiaque
chronique.27-30
Dans ces essais, le traitement à long terme par les antagonistes du
récepteur de l'endothéline n'a eu aucun effet sur le remodelage
ventriculaire gauche ou les résultats
cliniques.27-30
Conceptuellement, toutefois, l'administration à court terme
intraveineux d'un traitement peut avoir moins de possibilités de
montrer un effet à long terme sur la morbidité et la
mortalité, soulignant la difficulté d'évaluer de nouveaux
traitements dans l'insuffisance cardiaque aiguë. En dépit de ses
actions hémodynamiques, la milrinone n'a pas diminué la
morbidité et la mortalité dans l'essai OPTIME-CHF (Outcomes of a
Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart
Failure), et le nésiritide n'a pas diminué la moribidité
et la mortalité dans l'insuffisance cardiaque
aiguë.31-33
Naturellement, il est aussi possible qu'un effet bénéfique du
tezosentan dans un sous-groupe de patients puisse être
contrebalancé par un effet négatif dans un autre, par exemple,
les patients ayant une pression artérielle basse. VERITAS n'a pas,
cependant, eu la puissance statistique pour ce type d'analyse, ce qui, dans
tous les cas, ne pourrait que générer une hypothèse.
En résumé, le tezosentan, un traitement avec des actions
hémodynamiques « favorables », n'a pas réussi
à améliorer la dyspnée ou à diminuer les
événements cardio-vasculaires mortels ou non mortels chez des
patients admis en urgence pour insuffisance cardiaque aiguë. Il a
été impossible jusqu'à maintenant d'identifier un
rôle thérapeutique pour les antagonistes de l'endothéline
chez les patients ayant une insuffisance cardiaque aiguë ou
chronique.
Informations sur les auteurs
Correspondance: John J. V. McMurray, MD, Department of Cardiology,
Western Infirmary, Glasgow, G12 8QQ, United Kingdom
(j.mcmurray{at}bio.gla.ac.uk).
Les affiliations des auteurs et des investigateurs VERITAS sont
indiquées à la fin de cet article.
Contributions de l'auteur: Le Dr McMurray a eu un accès
complet à toutes les données de l'étude et accepte la
responsabilité de l'intégrité des données et de
l'exactitude de l'analyse des données.
Schéma et conception de l'étude: McMurray, Teerlink,
Cotter, Bourge, Cleland, Jondeau, Krum, Metra, O'Connor, Torre-Amione, van
Veldhuisen, Rainisio, Kobrin.
Recueil des données: McMurray, Teerlink, Cotter, Bourge,
Cleland, Jondeau, Krum, Parker, van Veldhuisen, Frey, Kobrin.
Analyse et interprétation des données: McMurray,
Teerlink, Cotter, Cleland, Krum, Metra, O'Connor, Parker, van Veldhuisen,
Lewsey, Rainisio, Kobrin.
Rédaction du manuscrit: McMurray, Teerlink, Cotter,
Cleland, Krum, Metra, O'Connor, Parker, van Veldhuisen.
Revue critique du manuscrit: McMurray, Teerlink, Cotter, Bourge,
Cleland, Jondeau, Krum, Metra,O'Connor, Torre-Amione, van Veldhuisen, Lewsey,
Frey, Rainisio, Kobrin.
Analyse statistique: Teerlink, Cotter, Lewsey, Rainisio.
Obtention du financement: McMurray.
Aide administrative, technique et matérielle: Teerlink,
Bourge, Cleland, Jondeau, Parker, van Veldhuisen, Frey.
Supervision de l'étude: McMurray, Teerlink, Cleland, Krum,
Metra, O'Connor, Parker, Torre-Amione, vanVeldhuisen.
Liens financiers: Aucun déclaré.
Financement/Soutien: Les essais VERITAS ont été
finances par Actelion Ltd. Les Drs Frey, Rainisio, et Kobrin travaillent chez
Actelion.
Rôle du sponsor: Actelion Ltd a conçu l'essai en
conjonction avec le comité scientifique VERITAS et a été
responsable des données de l'étude; mise à part la revue
par les Drs Frey et Kobrin, Actelion n'a joué aucun rôle dans la
préparation ou l'approbation du manuscrit.
Analyse statistique indépendante: Les données ont
été analysées par le sponsor et par un statisticien
indépendant (Dr Lewsey). Le Dr Lewsey a reçu une copie
complète de toutes les données et un plan analytique
prédéfinie; il a évalué le plan d'analyse pour son
exactitude en termes de réalisation des objectifs de l'étude et
il a mené une analyse statistique indépendante de toutes les
données, à l'aide du logiciel SAS version 9 (SAS Institute Inc).
Le Dr Lewsey a répliqué les résultats
présentés dans la section Résultats, les tableaux et
figures de cet article. Il y avait plusieurs différences mineures, qui
ont été résolues avec le statisticien du sponsor et les
résultats des analyses du Dr Lewsey reflètent celles
rapportées dans l'étude. La compensation financière du Dr
Lewsey a été fournie par l'University of Glasgow Discretionary
Research Fund.
Investigateurs VERITAS-1: Richard Pacher, University of Vienna,
Austria; Gunnar Jensen, Roskilde Amt Sygehus, Roskilde, Denmark; Karl Josef
Osterziel, Campus Virchow-Klinikum,Medizinische Klinik mit Schwerpunkt
Kardiologie, Berlin, Germany; Peter Hanrath, Medizinische Klinik I,
Universitatsklinikum Aachen, Aachen, Germany; Burkert Pieske,
Georg-August-Universitat Gottingen, Gottingen, Germany; John Nanas, University
of Athens, Greece; Guillaume Jondeau, Hopital Ambroise Pare, Boulogne, France;
Francois Funck, Heart Failure Clinic C. H. Dubos, Pontoise, France;
Gerald-Jose Roul, Hopitaux Universitaires de Strasbourg, Hopital Hautepierre,
Strasbourg, France; Michel Galinier, CHU Rangueil, Toulouse, France; Lucas
Liaudet, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland;
Tiziano Moccetti, Cardio Centro Ticino, Lugano, Switzerland; Marco Maggiorini,
University Hospital Zurich, Zurich, Switzerland; John Cleland, Hull Royal
Infirmary, Kingstonupon- Hull, United Kingdom; Jatinder Dhawan, Scunthorpe
General Hospital, Scunthorpe, United Kingdom; Theresa McDonagh and Mark
Petrie, Glasgow Royal Infirmary, Glasgow, United Kingdom; Reuven Zimlichmann,
Wolfson Medical Center, Holon, Israel; Gad Cotter, Assaf-Harofeh Medical
Center, Zriffin, Israel; Basil S. Lewis, Carmel Medical Center, Haifa, Israel;
Mohammed Omary, Nazareth Hopital EMMS, Nazareth, Israel; Leonardo Reisin,
Barzilai Hospital, Ashkelon, Israel; Chaim Lotan, Hadassah Hospital,
Jerusalem, Israel; Walentyna Mazurek, Katedra I Klinika Kardiologii AM,
Wroclaw, Poland; Kalina Kawecka- Jaszcz, Collegium Medicum UJ, Krakow, Poland;
Jacek Kubica, Samodzielny Publiczny Szpital, Bydgoszcz, Poland; Andrzej
Rynkiewicz, Akademia Medycznaw-Gdansku, Gdansk, Poland; Wieslawa Tracz,
College of Medicine of Jagellonian University, Krakow, Poland; Jacek
Gryzybowski, National Institute of Cardiology, Warsaw, Poland; John Parker,
Mount Sinai Hospital, Toronto, Ontario, Canada; Gordon Moe, St Michael's
Hospital, Toronto, Ontario, Canada; Anique Ducharme, Montreal Heart Institute,
Montreal, Quebec, Canada; Haissam Haddad, Ottawa Heart Institute,Ottawa,
Ontario, Canada; Serge Lepage, Hospital Fleurimont, Sherbrooke, Quebec,
Canada; J. Dale Cannon, Sumter Medical Consultants, Sumter, South Carolina;
Douglas Chapman, Heart Consultants, Omaha, Nebraska; Alan Maisel, VA San Diego
Healthcare System, San Diego, California; Reynolds Delgado, Texas Heart
Institute, Houston; Norma Keller, Bellevue Hospital, New York, New York;
Bradley Weinberg, Indiana Heart Hospital, Indianapolis; Bramah Singh, VA
Medical Center of West Los Angeles, Los Angeles, California; Lynne Wagoner,
University Hospital, Cincinnati, Ohio; David O. Taylor, Cleveland Clinic
Foundation, Cleveland, Ohio; Jalal Ghali, LSU Health Sciences Center,
Shreveport, Louisiana; John Teerlink, Department of Veterans Affairs, San
Francisco, California; Denise Hermann, UCSD Medical Center, San Diego,
California; Biykem Bozkurt, Veterans Affairs Medical Center, Houston, Texas;
T. Barry Levine, Heart Hospital at Allegheny General, Pittsburgh,
Pennsylvania; Jose A. Talíaj, University of Alabama-Birmingham; Brack
Hattler, Denver Veterans Affairs Medical Center, Denver, Colorado; Thomas
Amidon, Medical Associates, Bellevue, Washington; Frances Johnson and John
Giacomini, Palo Alto VA Healthcare System, Palo Alto, California; Thadius
Tolleson, Tyler Cardiovascular Consultants, Tyler, Texas; Boshara Zakhary,
Danville Medical Center, Danville, Virginia.
Investigateurs VERITAS-2: Karl Stangl, Universitatsklinikum der
Humboldt-Universitat Berlin, Berlin, Germany; Stephan B. Felix, Universitat
Greifswald, Greifswald, Germany; Christian Hengstenberg, Klinik u. Poliklinik
f. Inn. Med. II, Regensburg, Germany; Hans-Georg Olbrich, Asklepios Klinik
Langen, Langen, Germany; Franz Hartmann, Universitatsklinikum Schleswig
Holstein, Lubeck, Germany; Livio Dei Cas and Marco Metra, Cattedra di
Cardiologia, Brescia, Italy; Dan Atar, Aker University Hospital, Oslo, Norway;
Kenneth Dickstein, Central Hospital in Rogaland, Stavanger, Norway; Torstein
Hole, Sentralsykehuset IMore og Romsdal, Alesund, Norway; Edoardo Gronda,
Istituto Clinico Humanitas, Rozzano, Italy; John McMurray, University of
Glasgow West, Glasgow, United Kingdom; Iain Squire, University of Leicester,
Leicester, United Kingdom; Robert MacFayden, City Hospital, Birmingham, United
Kingdom; M. A. Memon, Bridlington & District Hospital, Bridlington, United
Kingdom; Roxy Senior, Northwick Park Hospital, Harrow, United Kingdom; Edo
Kaluski, Assaf-Harofe Hospital, Zriffin, Israel; Doron Zahger, Soroka
University Medical Center, Beer-Sheva, Israel; Alon Marmor, Rebecca-Zieff
Medical Center, Safed, Israel; Abraham Caspi, Kaplan Hospital, Rehovot,
Israel; N. Rougin, Western Galilee Hospital, Nahariya, Israel; Teddy Abraham
Weiss, Hadassah University Hospital, Jerusalem, Israel; Dov Gavish, E. Wolfson
Medical Center, Holon, Israel; D. J. van Veldhuisen, University Hospital
Groningen, Groningen, the Netherlands; P.M. Landsaat and J. P. R.
Hermann, OLVG, Cardio Research, Amsterdam, the Netherlands; G. L. Bartels,
Cardio Research Noord bv, Groningen, the Netherlands; H. R. Michels, Catharina
Ziekenhuis Eindhoven, Eindhoven, the Netherlands; B. J. W. M. Rensing,
Stichting Sint Antonius Ziekenhuis, Nieuwegein, the Netherlands; Duc Nam
Nguyen, Akademisch Ziekenhuis, Brussels, Belgium; Martin Herold, University
Hospital Vinohrady, Prague, Czech Republic; Bronislav Janek, Klinika
Kardiologie IKEM, Prague, Czech Republic; Miroslav Soucek, Faculty Hospital St
Anna, Brno, Czech Republic; Frantisek Holm, Krajska Nemocnice Liberec,
Liberec, Czech Republic; Iveta Petrova, Masaryk Hospital, Usti nad Labem,
Czech Republic; IstvanE des, University of Debrecen, Debrecen, Hungary;
Tamá s Forster, 2nd Department of Medicine & Cardiology Centre,
Szeged, Hungary; Gyorgy Sallai, Delpesti Korhaz, Budapest, Hungary; Janos
Tomcsanyi, Polyclinic of the Hospitaler Brothers of St John of God, Budapest,
Hungary; Henry Krum, Monash University and Alfred Hospital, Prahran, Victoria,
Australia; John Horowitz, Queen Elizabeth Hospital, Woodville, South
Australia, Australia; Andrew Sindone, Concord Repatriation Hospital, Concord,
New South Wales, Australia; Bleakley Chandler, University Hospital, Augusta,
Georgia; David Rubinstein, Elmhurst Hospital Center, Elmhurst, New York; Uri
Elkayam, LA County-USC Medical Center, Los Angeles, California; Ron Oren,
University of Iowa Hospital and Clinics, Iowa City; Rafael Sequeira,
University of Miami-Jackson Memorial Hospital, Miami, Florida; Monica Shah and
Christopher O'Connor, Duke University Medical Center, Durham, North Carolina;
Guillermo Torre, Baylor College of Medicine, Houston, Texas; Ignatius Thomas,
Medical Research Institute, Slidell, Louisiana; Deborah Ascheim, Heart Failure
Center at Columbia Presbyterian Medical Center, New York, New York; Kirkwood
Adams, University of North Carolina, Chapel Hill; Jake Hochrein, LeBauer
Cardiovascular Research Foundation, Greensboro, North Carolina; Michael J.
Koren, Jacksonville Center for Clinical Research, Jacksonville, Florida; Mara
T. Slawsky, Baystate Medical Center, Springfield, Massachusetts; Ulrich P.
Jorde, New York University School of Medicine, New York; W. Herbert Haught,
Oracle Research, Huntsville, Alabama; Daniel J. Lenihan, M.D. Anderson Cancer
Center, Houston, Texas. and J. P. R.
Hermann, OLVG, Cardio Research, Amsterdam, the Netherlands; G. L. Bartels,
Cardio Research Noord bv, Groningen, the Netherlands; H. R. Michels, Catharina
Ziekenhuis Eindhoven, Eindhoven, the Netherlands; B. J. W. M. Rensing,
Stichting Sint Antonius Ziekenhuis, Nieuwegein, the Netherlands; Duc Nam
Nguyen, Akademisch Ziekenhuis, Brussels, Belgium; Martin Herold, University
Hospital Vinohrady, Prague, Czech Republic; Bronislav Janek, Klinika
Kardiologie IKEM, Prague, Czech Republic; Miroslav Soucek, Faculty Hospital St
Anna, Brno, Czech Republic; Frantisek Holm, Krajska Nemocnice Liberec,
Liberec, Czech Republic; Iveta Petrova, Masaryk Hospital, Usti nad Labem,
Czech Republic; IstvanE des, University of Debrecen, Debrecen, Hungary;
Tamá s Forster, 2nd Department of Medicine & Cardiology Centre,
Szeged, Hungary; Gyorgy Sallai, Delpesti Korhaz, Budapest, Hungary; Janos
Tomcsanyi, Polyclinic of the Hospitaler Brothers of St John of God, Budapest,
Hungary; Henry Krum, Monash University and Alfred Hospital, Prahran, Victoria,
Australia; John Horowitz, Queen Elizabeth Hospital, Woodville, South
Australia, Australia; Andrew Sindone, Concord Repatriation Hospital, Concord,
New South Wales, Australia; Bleakley Chandler, University Hospital, Augusta,
Georgia; David Rubinstein, Elmhurst Hospital Center, Elmhurst, New York; Uri
Elkayam, LA County-USC Medical Center, Los Angeles, California; Ron Oren,
University of Iowa Hospital and Clinics, Iowa City; Rafael Sequeira,
University of Miami-Jackson Memorial Hospital, Miami, Florida; Monica Shah and
Christopher O'Connor, Duke University Medical Center, Durham, North Carolina;
Guillermo Torre, Baylor College of Medicine, Houston, Texas; Ignatius Thomas,
Medical Research Institute, Slidell, Louisiana; Deborah Ascheim, Heart Failure
Center at Columbia Presbyterian Medical Center, New York, New York; Kirkwood
Adams, University of North Carolina, Chapel Hill; Jake Hochrein, LeBauer
Cardiovascular Research Foundation, Greensboro, North Carolina; Michael J.
Koren, Jacksonville Center for Clinical Research, Jacksonville, Florida; Mara
T. Slawsky, Baystate Medical Center, Springfield, Massachusetts; Ulrich P.
Jorde, New York University School of Medicine, New York; W. Herbert Haught,
Oracle Research, Huntsville, Alabama; Daniel J. Lenihan, M.D. Anderson Cancer
Center, Houston, Texas.
Affiliations des auteurs: Department of Cardiology, Western
Infirmary, Glasgow, United Kingdom; Section of Cardiology, San Francisco
Veterans Affairs Medical Center et University of California, San Francisco;
Division of Cardiology, Department of Medicine, Duke University Medical Center
et Duke Clinical Research Institute, Durham, North Carolina; Department of
Medicine, University of Alabama at Birmingham; Department of Cardiology,
Castlehill Hospital, University of Hull, Kingston upon Hull, United Kingdom;
Department of Cardiology, Hôpital Ambroise Paré, Boulogne,
France; Departments of Medicine, Epidemiology, et Preventive Medicine, Monash
University Central et Eastern Clinical School, Alfred Hospital, Melbourne,
Victoria, Australia; Section of Cardiovascular Diseases, Department of
Experimental and Applied Medicine, University of Brescia, Brescia, Italy;
Mount Sinai et University Health Network Hospitals, Toronto, Ontario, Canada;
Methodist DeBakey Heart Center,Winters Center for Heart Failure Research,
Baylor College of Medicine, Houston, Texas; Department of Cardiology, Thorax
Center, University Hospital Groningen, Groningen, the Netherlands; Department
of Public Health and Health Policy, University of Glasgow, Glasgow, United
Kingdom; Actelion Pharmaceuticals Ltd, Allschwil, Switzerland.
BIBLIOGRAPHIE
| |
1. Nieminen MS, Harjola VP. Definition and epidemiology of acute heart
failure syndromes. Am J Cardiol.2005; 96(6A):5G
-10G.
PUBMED
2. Zannad F, Adamopoulos C, Mebazaa A, Gheorghiade M. The challenge of
acute decompensated heart failure. Heart Fail Rev.2006; 11(2):135
-139.
PUBMED
3. Gheorghiade M, Zannad F, Sopko G, et al; International Working
Group on Acute Heart Failure Syndromes. Acute heart failure syndromes: current
state and framework for future research. Circulation.2005; 112(25):3958
-3968.
FREE FULL TEXT
4. Sharma M, Teerlink JR. A rational approach for the treatment of
acute heart failure: current strategies and future options. Curr
Opin Cardiol.2004; 19(3):254
-263.
PUBMED
5. Nieminen MS, Bohm M, Cowie MR, et al; ESC Committee for Practice
Guideline (CPG). Executive summary of the guidelines on the diagnosis and
treatment of acute heart failure: the Task Force on Acute Heart Failure of the
European Society of Cardiology. Eur Heart J.2005; 26(4):384
-416.
FREE FULL TEXT
6. McMurray JJ, Ray SG, Abdullah I, Dargie HJ, Morton JJ. Plasma
endothelin in chronic heart failure. Circulation.1992; 85(4):1374
-1379.
FREE FULL TEXT
7. Van Beneden R, Gurne O, Selvais PL, et al. Superiority of big
endothelin-1 and endothelin-1 over natriuretic peptides in predicting survival
in severe congestive heart failure: a 7-year follow-up study. J
Card Fail.2004; 10(6):490
-495.
PUBMED
8. Masson S, Latini R, Anand IS, et al; Val-HeFT investigators. The
prognostic value of big endothelin-1 in more than 2,300 patients with heart
failure enrolled in the Valsartan Heart Failure Trial (Val-HeFT). J
Card Fail.2006; 12(5):375
-380.
PUBMED
9. Torre-Amione G, Young JB, Durand J, et al. Hemodynamic effects of
tezosentan, an intravenous dual endothelin receptor antagonist, in patients
with class III to IV congestive heart failure.
Circulation.2001; 103(7):973
-980.
FREE FULL TEXT
10. Kaluski E, Kobrin I, Zimlichman R, et al. RITZ-5: Randomized
Intravenous TeZosentan (an endothelin-A/B antagonist) for the treatment of
pulmonary edema: a prospective, multicenter, double-blind, placebo-controlled
study. J Am Coll Cardiol.2003; 41(2):204
-210.
FREE FULL TEXT
11. O'Connor CM, Gattis WA, Adams KF Jr, et al; Randomized Intravenous
TeZosentan Study-4 Investigators. Tezosentan in patients with acute heart
failure and acute coronary syndromes: results of the Randomized Intravenous
TeZosentan Study (RITZ-4). J Am Coll Cardiol.2003; 41(9):1452
-1457.
FREE FULL TEXT
12. Torre-Amione G, Young JB, Colucci WS. Hemodynamic and clinical
effects of tezosentan, an intravenous dual endothelin receptor antagonist, in
patients hospitalized for acute decompensated heart failure. J Am
Coll Cardiol.2003; 42(1):140
-147.
FREE FULL TEXT
13. Cotter G, Kaluski E, Stangl K, et al. The hemodynamic and
neurohormonal effects of low doses of tezosentan (an endothelin A/B receptor
antagonist) in patients with acute heart failure. Eur J Heart
Fail.2004; 6(5):601
-609.
FREE FULL TEXT
14. Teerlink JR, McMurray JJ, Bourge RC, et al; VERITAS Investigators.
Tezosentan in patients with acute heart failure: design of the Value of
Endothelin Receptor Inhibition with Tezosentan in Acute heart failure Study
(VERITAS). Am Heart J.2005; 150(1):46
-53.
PUBMED
15. Slawsky MT, Colucci WS, Gottlieb SS, et al. Acute hemodynamic and
clinical effects of levosimendan in patients with severe heart failure.
Circulation.2000; 102(18):2222
-2227.
FREE FULL TEXT
16. Colucci WS, Elkayam U, Horton DP, et al; Nesiritide Study Group.
Intravenous nesiritide, a natriuretic peptide, in the treatment of
decompensated congestive heart failure. N Engl J Med.2000; 343(4):246
-253.
PUBMED
17. Louis A, Cleland JG, Crabbe S, et al. Clinical trials update:
CAPRICORN, COPERNICUS, MIRACLE, STAF, RITZ-2, RECOVER and RENAISSANCE and
cachexia and cholesterol in heart failure: Highlights of the Scientific
Sessions of the American College of Cardiology, 2001. Eur J Heart
Fail.2001; 3(3):381
-387.
FREE FULL TEXT
18. Teerlink JR. Dyspnea as an end point in clinical trials of
therapies for acute decompensated heart failure. Am Heart
J.2003; 145(2)(suppl):S26
-S33.
PUBMED
19. Teerlink JR, Massie BM, Cleland JGF, Tzivoni D; for the RITZ-1
Investigators. A double-blind, parallel-group, multi-center,
placebo-controlled study to investigate the efficacy and safety of tezosentan
in reducing symptoms in patients with acute decompensated heart failure
[abstract]. Circulation.2001; 104:526
.
20. Moiseyev VS, Poder P, Andrejevs N, et al; RUSSLAN Study
Investigators. Safety and efficacy of a novel calcium sensitizer,
levosimendan, in patients with left ventricular failure due to an acute
myocardial infarction: a randomized, placebo-controlled, double-blind study
(RUSSLAN). Eur Heart J.2002; 23(18):1422
-1432.
FREE FULL TEXT
21. Publication Committee for the VMAC Investigators (Vasodilatation in
the Management of Acute CHF). Intravenous nesiritide vs nitroglycerin for
treatment of decompensated congestive heart failure: a randomized controlled
trial. JAMA.2002; 287(12):1531
-1540.
FREE FULL TEXT
22. US Food and Drug Administration. NDA 20-920 Natrecor (nesiritide).
http://www.fda.gov/ohrms/dockets/ac/01/briefing/3749b2_02_02-FDA-Medical%20Review.pdf.
Accessibility verified October 12, 2007.23. Cleland JG, Freemantle N, Coletta AP, Clark AL. Clinical trials
update from the American Heart Association: REPAIR-AMI, ASTAMI, JELIS, MEGA,
REVIVE-II, SURVIVE, and PROACTIVE. Eur J Heart Fail.2006; 8(1):105
-110.
PUBMED
24. Gheorghiade M, Konstam MA, Burnett JC Jr, et al; Efficacy of
Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST)
Investigators. Short-term clinical effects of tolvaptan, an oral vasopressin
antagonist, in patients hospitalized for heart failure: the EVEREST clinical
status trials. JAMA.2007; 297(12):1332
-1343.
FREE FULL TEXT
25. Binanay C, Califf RM, Hasselblad V, et al; ESCAPE Investigators and
ESCAPE Study Coordinators. Evaluation study of congestive heart failure and
pulmonary artery catheterization effectiveness: the ESCAPE trial.
JAMA.2005; 294(13):1625
-1633.
FREE FULL TEXT
26. Grant S, Aitchison T, Henderson E, et al. A comparison of the
reproducibility and the sensitivity to change of visual analogue scales, Borg
scales, and Likert scales in normal subjects during submaximal exercise.
Chest.1999; 116(5):1208
-1217.
PUBMED
27. Anand I, McMurray J, Cohn JN, et al; EARTH Investigators. Long-term
effects of darusentan on left-ventricular remodelling and clinical outcomes in
the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH):
randomised, double-blind, placebo-controlled trial.
Lancet.2004; 364(9431):347
-354.
PUBMED
28. Packer M, McMurray J, Massie BM, et al. Clinical effects of
endothelin receptor antagonism with bosentan in patients with severe chronic
heart failure: results of a pilot study. J Card Fail.2005; 11(1):12
-20.
PUBMED
29. Coletta A, Thackray S, Nikitin N, Cleland JG. Clinical trials
update: highlights of the scientific sessions of the American College of
Cardiology 2002: LIFE, DANAMI 2, MADIT-2, MIRACLE-ICD, OVERTURE, OCTAVE,
ENABLE 1 & 2, CHRISTMAS, AFFIRM, RACE, WIZARD, AZACS, REMATCH, BNP trial
and HARDBALL. Eur J Heart Fail.2002; 4(3):381
-388.
FREE FULL TEXT
30. Prasad SK, Dargie HJ, Smith GC, et al. Comparison of the dual
receptor endothelin antagonist enrasentan with enalapril in asymptomatic left
ventricular systolic dysfunction: a cardiovascular magnetic resonance study.
Heart.2006; 92(6):798
-803.
FREE FULL TEXT
31. Cuffe MS, Califf RM, Adams KF Jr, et al; Outcomes of a Prospective
Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure
(OPTIME-CHF) Investigators. Short-term intravenous milrinone for acute
exacerbation of chronic heart failure: a randomized controlled trial.
JAMA.2002; 287(12):1541
-1547.
FREE FULL TEXT
32. Sackner-Bernstein JD, Skopicki HA, Aaronson KD. Risk of worsening
renal function with nesiritide in patients with acutely decompensated heart
failure. Circulation.2005; 111(12):1487
-1491.
FREE FULL TEXT
33. Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term
risk of death after treatment with nesiritide for decompensated heart failure:
a pooled analysis of randomized controlled trials.
JAMA.2005; 293(15):1900
-1905.
FREE FULL TEXT
ARTICLE EN RAPPORT
Cette semaine dans le JOURNAL
JAMA. 2007;298:1977.
Texte Complet
|