|
Show all abstracts Show selected abstracts Add to my list |
|
| EDITORIAL |
|
|
|
Warfarin pharmacogenetics: How close are we to clinical practice? |
p. 277 |
Tejasvita Gaikwad, Shrimati Shetty, Kanjaksha Ghosh
DOI:10.4103/0971-6866.120806 PMID:24339537 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| REVIEW ARTICLES |
 |
|
|
|
Hemoglobin E disorder: Newborn screening program |
p. 279 |
Viroj Wiwanitkit
DOI:10.4103/0971-6866.120808 PMID:24339538Hemoglobin E (Hb E) disorder is an important kind of hemoglobinopathy. It can be seen around the world with the highest prevalence in Southeast Asia. The screening for this disorder becomes the public health policies in many countries. The screening can be performed in several population groups. The newborn screening program for Hb E disorder is an important issue in pediatric genetics. In this brief review, the author discusses on important laboratory tests for screening for Hb E disorder in newborn. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
An overview of gene therapy in head and neck cancer |
p. 282 |
Amit Bali, Deepika Bali, Ashutosh Sharma
DOI:10.4103/0971-6866.120811 PMID:24339539Gene therapy is a new treatment modality in which new gene is introduced or existing gene is manipulated to cause cancer cell death or slow the growth of the tumor. In this review, we have discussed the different treatment approaches for cancer gene therapy; gene addition therapy, immunotherapy, gene therapy using oncolytic viruses, antisense ribonucleic acid (RNA) and RNA interference-based gene therapy. Clinical trials to date in head and neck cancer have shown evidence of gene transduction and expression, mediation of apoptosis and clinical response including pathological complete responses. The objective of this article is to provide an overview of the current available gene therapies for head and neck cancer. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| ORIGINAL ARTICLES |
|
|
|
|
Usage of U7 small nuclear ribonucleic acid in gene therapy of hemoglobin D Punjab disorder: Rationale? |
p. 291 |
Viroj Wiwanitkit
DOI:10.4103/0971-6866.120812 PMID:24339540Background: Hemoglobin (Hb) D Punjab disorder is a congenital hemoglobinopathy described in India. It is a disorder due to defect in beta-globin gene.
Materials and Methods: Here, the author assesses the possibility of U7.623 gene therapy for Hb D Punjab disorder. A standard bioinformatic analysis to study the effect of co-expression between nucleic acid sequence for human Hb D Punjab beta-globin chain and U7.623 was performed.
Result: It can be seen that fully recovery of Hb function and biological process can be derived via gene ontology study.
Conclusion: Here, there is a rationale to use U7 small nuclear ribonucleic acid as a possible tool for gene therapy in Hb D Punjab disorder. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
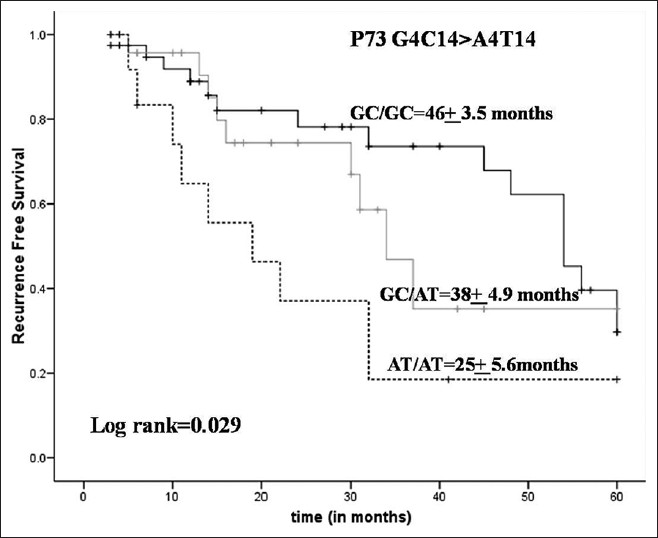  |
Polymorphism at P21 codon 31 and dinucleotide polymorphism of P73 gene and susceptibility to bladder cancer in individuals from North India |
p. 293 |
Praveen Kumar Jaiswal, Vibha Singh, Rama Devi Mittal
DOI:10.4103/0971-6866.120815 PMID:24339541Background and Aim: p73, a novel P53 homolog and plays an important role in modulating cell cycle control, apoptosis and cell growth while P21, functions to negatively control the cell cycle. P53 up regulates p21 expression in response to deoxyribonucleic acid damage leading to cell cycle arrest at G1 checkpoint. In the present study, we are targeting p21 codon 31 and p73 gene variants of G4C14-to-A4T14 (Exon 2) polymorphism for bladder cancer (BC) risk in North Indians.
Materials and Methods: The above gene variants of P21 and P73 were assessed in the case-control study comprising of 200 BC cases and 200 healthy controls of the same age, gender and similar ethnicity. Genotyping was performed by polymerase chain reaction (PCR) restriction fragment length polymorphism method and PCR-based confronting two-pair primers (PCR with CTPP).
Results: The variant genotype of p73Exon 2 polymorphism showed significant risk for BC (p = 0.014). While combining with heterozygous genotype, variant genotype of p73Exon2 showed a significant association with BC risk (p = 0.010). While in case of p21 codon31 showed no significant association for BC risk at genotypic level. Significant association between p73Exon2 polymorphism and smoking was observed for BC risk. Furthermore, gene combination analysis revealed that AT/AT-Ser/Ser is associated with risk for BC. Variant genotype of P73Exon2 was associated with reduced risk of recurrence (p = 0.039) in superficial BC patients receiving Bacillus Calmette-Guerin treatment thus showing least survival (log rank = 0.029).
Conclusion: Our study provided evidence that the p73 G4C14 > A4T14 (Exon2) polymorphisms were associated with higher risk of BC in North Indian population. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
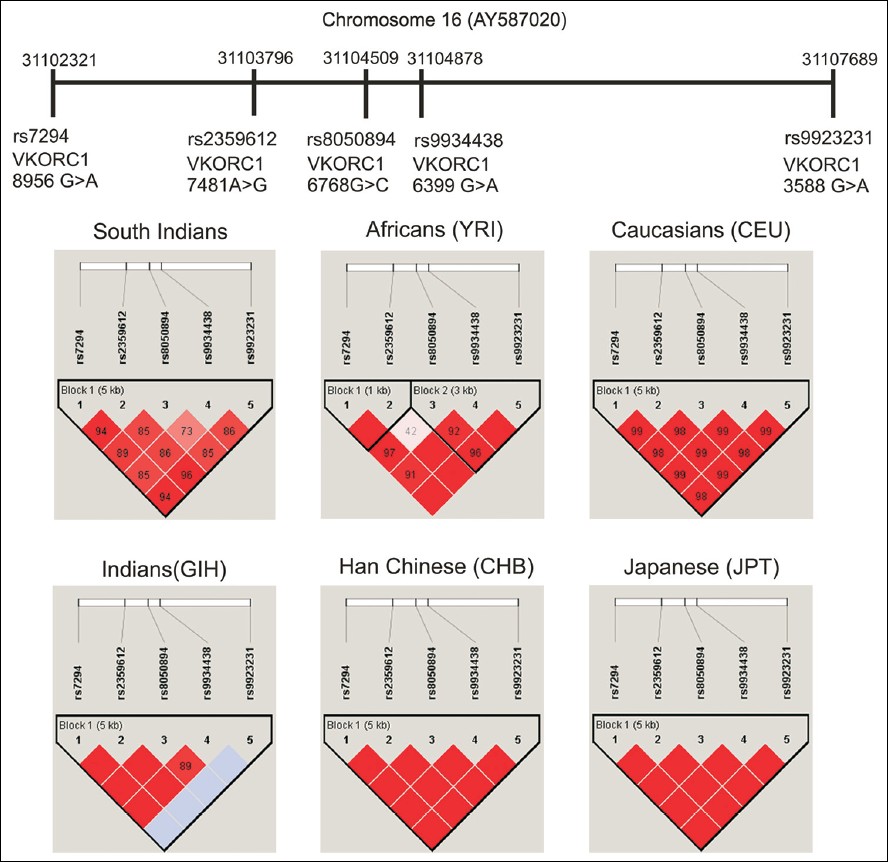 |
Inter and intra ethnic variation of vitamin K epoxide reductase complex and cytochrome P450 4F2 genetic polymorphisms and their prevalence in South Indian population |
p. 301 |
Dhakchinamoorthi Krishna Kumar, Deepak Gopal Shewade, Sajjanavar Manjunath, Prayaga Ushakiran, Gangadharan Reneega, Chandrasekaran Adithan
DOI:10.4103/0971-6866.120817 PMID:24339542Background: Genetic variation in the vitamin K epoxide reductase complex (VKORC1) and cytochrome P450 4F2 (CYP4F2) genes were found to be strongly associated with the oral anticoagulant (OA) dose requirement. The distribution of genetic variation in these two genes was found to show large inter- and intra-ethnic difference.
Materials and Methods: A total of 470 unrelated, healthy volunteers of South Indians of either sex (age: 18-60 years) were enrolled for the study. A 5 ml of venous blood was collected and the genomic deoxyribonucleic acid (DNA) was extracted by using phenol-chloroform extraction method. Real-time quantitative polymerase chain reaction (RT-PCR) method was used for genotyping.
Results: The variant allele frequencies of VKORC1 rs2359612 (T), rs8050894 (C), rs9934438 (T) and rs9923231 (A) were found to be 11.0%, 11.8%, 11.7% and 12.0%, respectively. The variant allele VKORC1 rs7294 was (80.1%) more frequent and the variant allele CYP4F2 * 3 was found to be 41.8% in South Indians. The allele, genotype and haplotype frequencies of VKORC1 and CYP4F2 gene were distinct from other compared HapMap populations (P < 0.0001).
Conclusion: The findings of our study provide the basic genetic information for further pharmacogenetic based investigation of OA therapy in the population. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (1) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
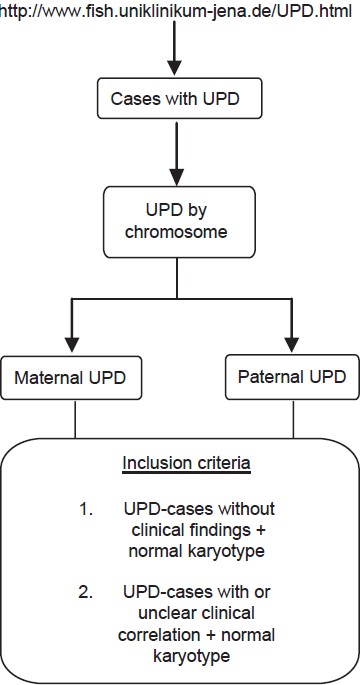 |
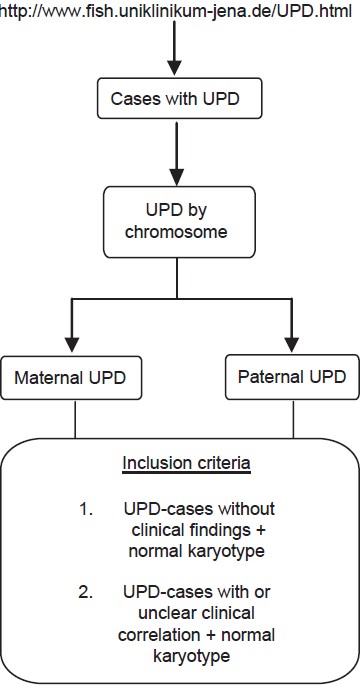
Phenotypic spectrum in uniparental disomy: Low incidence or lack of study? |
p. 311 |
Arpan D Bhatt, Thomas Liehr, Sonal R Bakshi
DOI:10.4103/0971-6866.120819 PMID:24339543Context: Alterations in the human chromosomal complement are expressed phenotypically ranging from (i) normal, via (ii) frequent fetal loss in otherwise normal person, to (iii) sub-clinical to severe mental retardation and dysmorphism in live births. A subtle and microscopically undetectable chromosomal alteration is uniparental disomy (UPD), which is known to be associated with distinct birth defects as per the chromosome involved and parental origin. UPD can be evident due to imprinted genes and/or activation of recessive mutations.
Aims: The present study comprises of data mining of published UPD cases with a focus on associated phenotypes. The goal was to identify non-random and recurrent associations between UPD and various genetic conditions, which can possibly indicate the presence of new imprinted genes.
Settings and Design: Data mining was carried out using the homepage "http://www.fish.uniklinikum-jena.de/UPD.html," an online catalog of published cases with UPD.
Materials and Methods: The UPD cases having normal karyotype and with or without clinical findings were selected to analyze the associated phenotypes for each chromosome, maternal or paternal involved in UPD.
Results: Our results revealed many genetic conditions (other than the known UPD syndromes) to be associated with UPD. Even in cases of bad obstetric history as well as normal individuals chance detection of UPD has been reported.
Conclusions: The role of UPD in human genetic disorders needs to be studied by involving larger cohorts of individuals with birth defects as well as normal population. The genetic conditions were scrutinized in terms of inheritance patterns; majority of these were autosomal recessive indicating the role of UPD as an underlying mechanism. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Index of opportunity for natural selection among the Gowdas of Kodagahalli village, Karnataka, India |
p. 315 |
Bhaboklang Sohkhlet
DOI:10.4103/0971-6866.120821 PMID:24339544Background: In order to understand how selection is operating in the Gowda population, the index of opportunity for selection was calculated and the present findings were compared with some related findings from other South Indian (SI) populations.
Materials and Methods: Crow (1958) and the modified method by Johnston and Kensinger (1971) were used for the present purpose.
Results and Discussion: The index of total selection intensity (I) was found to be moderate taking into consideration the range for many Indian populations. Considering certain differences in fertility and mortality heritable, it appears that natural selection play an important role in shaping the genetic constitution of the Gowda population. Analysis of data indicates that the index due to fertility seems to contribute more towards selection than mortality. This trend might be because of better living condition and health-care system among the Gowdas which have a positive impact on the lower contribution of mortality for the evolution mechanism of the Gowda population through natural selection. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
No CAG repeat expansion of polymerase gamma is associated with male infertility in Tamil Nadu, South India |
p. 320 |
J Poongothai
DOI:10.4103/0971-6866.120823 PMID:24339545Mitochondria contains a single deoxyribonucleic acid (DNA) polymerase, polymerase gamma (POLG) mapped to long arm of chromosome 15 (15q25), responsible for replication and repair of mitochondrial DNA. Exon 1 of the human POLG contains CAG trinucleotide repeat, which codes for polyglutamate. Ten copies of CAG repeat were found to be uniformly high (0.88) in different ethnic groups and considered as the common allele, whereas the mutant alleles (not -10/not -10 CAG repeats) were found to be associated with oligospermia/oligoasthenospermia in male infertility. Recent data suggested the implication of POLG CAG repeat expansion in infertility, but are debated. The aim of our study was to explore whether the not -10/not -10 variant is associated with spermato g enic failure. As few study on Indian population have been conducted so far to support this view, we investigated the distribution of the POLG CAG repeats in 61 infertile men and 60 normozoospermic control Indian men of Tamil Nadu, from the same ethnic background. This analysis interestingly revealed that the homozygous wild type genotype (10/-10) was common in infertile men (77% - 47/61) and in normozoospermic control men (71.7% - 43/60). Our study failed to confirm any influence of the POLG gene polymorphism on the efficiency of the spermatogenesis. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Linkage study of DFNB3 responsible for hearing loss in human |
p. 325 |
Akhtar Ali, Masroor E Babar, Saeeda Kalsoom, Jamil Ahmad, Kamran Abbas
DOI:10.4103/0971-6866.120827 PMID:24339546Background: Hearing disorders represent a significant health problem worldwide. Recessive inherited cases of the deafness are more prevalent in Pakistan due to consanguineous marriages. Deafness caused by DFNB3 is due to mutation in the gene MYO XVA and its prevalence among Pakistani population is about 5%.
Materials and Methods: Families with at least two or more individual affected with deafness were selected from different areas of District Okara of Pakistan. Six consanguineous families of different ethnic groups having deaf individuals were studied. All these families had three or more deaf individuals in either two or more sib ships. Family history was taken to minimize the chances of other abnormalities. Pedigrees drawn by using Cyrillic software (version 2.1) showed that all the marriages were consanguineous and the families have recessive mode of inheritance. Three STR markers were selected and amplified on all the samples of six families through PCR. The PCR products were then genotyped on non denaturing polyacrylamide gel electrophoresis (PAGE). Haplotypes were constructed to determine the pattern of inheritance and also to determine whether a family was linked or unlinked with known DFNB3 locus.
Results: One out of six families showed linkage to the DFNB3 while rest of the families remained unlinked. Carriers of deafness genes were identified and information was provided to the families on request.
Conclusion: Knowledge about the genetic causes of deafness provide insight into the variable expression of genes involved in this hereditary problem and may allow the prediction and prevention of associated health problems. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
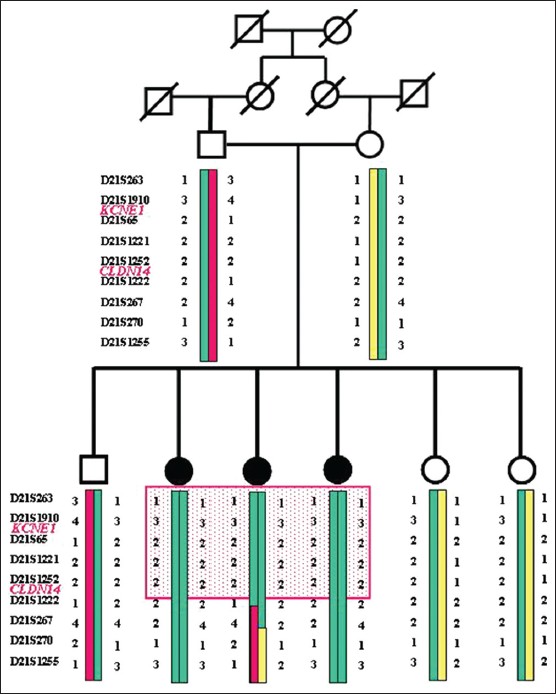 |
Genetic and molecular analysis of the CLDN14 gene in Moroccan family with non-syndromic hearing loss |
p. 331 |
Majida Charif, Redouane Boulouiz, Amina Bakhechane, Houda Benrahma, Halima Nahili, Abdelmajid Eloualid, Hassan Rouba, Mostafa Kandil, Omar Abidi, Guy Lenaers, Abdelhamid Barakat
DOI:10.4103/0971-6866.120828 PMID:24339547Background: Hearing loss is the most prevalent human genetic sensorineural defect. Mutations in the CLDN14 gene, encoding the tight junction claudin 14 protein expressed in the inner ear, have been shown to cause non-syndromic recessive hearing loss DFNB29.
Aim: We describe a Moroccan SF7 family with non-syndromic hearing loss. We performed linkage analysis in this family and sequencing to identify the mutation causing deafness.
Materials and Methods: Genetic linkage analysis, suggested the involvement of CLDN14 and KCNE1 gene in deafness in this family. Mutation screening was performed using direct sequencing of the CLDN14 and KCNE1 coding exon gene.
Results: Our results show the presence of c.11C>T mutation in the CLDN14 gene. Transmission analysis of this mutation in the family showed that the three affected individuals are homozygous, whereas parents and three healthy individuals are heterozygous. This mutation induces a substitution of threonine to methionine at position 4.
Conclusion: These data show that CLDN14 gene can be i mplicated in the development of hearing loss in SF7 family; however, the pathogenicity of c.11C>T mutation remains to be determined. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Analysis of hemoglobin electrophoresis results and physicians investigative practices in Saudi Arabia |
p. 337 |
Syed Riaz Mehdi, Badr Abdullah Al Dahmash
DOI:10.4103/0971-6866.120829 PMID:24339548Background and Objectives: Riyadh and central province falls in a moderate prevalent zone of hemoglobinopathies in Saudi Arabia. However, it has been observed that the physicians working in Saudi Arabia invariably advise all cases of anemia for hemoglobin electrophoresis (HE). The present work was carried out to study the yield of the HE in Riyadh and the investigative practices of the physicians advising HE.
Settings and Design: The study was carried out in the hospitals of King Saud University from 2009 to 2011 in order to assess the yield of HE in referred cases of clinical anemia.
Materials and Methods: A total of 1073 cases divided in two groups of males and females had undergone complete blood count and red blood cell morphology. Cellulose acetate HE was performed and all the positive results were reconfirmed on the high performance liquid chromatography (HPLC). The results were analyzed for the type of hemoglobinopathies. For statistical analysis Statistical Package for Social Sciences 15 version (SPSS Inc., Chicago, IL, USA) was used.
Results: A total of 405 males and 668 females blood samples were included in the present study. 116 (28.5%) males and 167 (25%) females showed an abnormal pattern on HE. The incidence of beta thalassemia trait was higher in females while sickle cell trait was predominantly seen in males. Red cell indices were reduced considerably in thalassemias, but were unaffected in sickle cell disorders, except those which had concurrent alpha trait. The total yield of HE was 26.6% which was much less than expected.
Conclusion: The physicians are advised to rule out iron deficiency and other common causes of anemia before investigating the cases for hemoglobinopathies, which employs time consuming and expensive tests of HE and HPLC. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Population based family history analysis of Brahmins in a small town in India for the prevalence of type-2 diabetes mellitus |
p. 342 |
Arun V Panat, Dilip A Kulkarni, Ravindra B Ghooi
DOI:10.4103/0971-6866.120826 PMID:24339549Objectives: The objective of this study is to determine the inheritance pattern of type-2 diabetes and make stratification for the general population risk.
Materials and Methods: A questionnaire was developed for o btaining the family history. Analysis of the data was carried out by using student and Chi-square tests and for stratification; the guidelines of Scheuner et al. were followed.
Results: The pattern of inheritance is the male sex specific (χ² =13.44). The mean age of onset of diabetes in parents was 58.61 ± 2.94 and in offspring 46.75 ± 2.54. In all 47.22 ± 11.53% families were found in high risk and 31.94 ± 10.77% in the moderate risk category. In female diabetics, the onset was in the age range of 41-60 years.
Conclusion: We found a high-risk of diabetes and familial clustering in successive generations of Brahmins with prominent male sex specificity. In females onset of diabetes was coinciding with the period around menopause. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| CASE REPORTS |
|
|
|
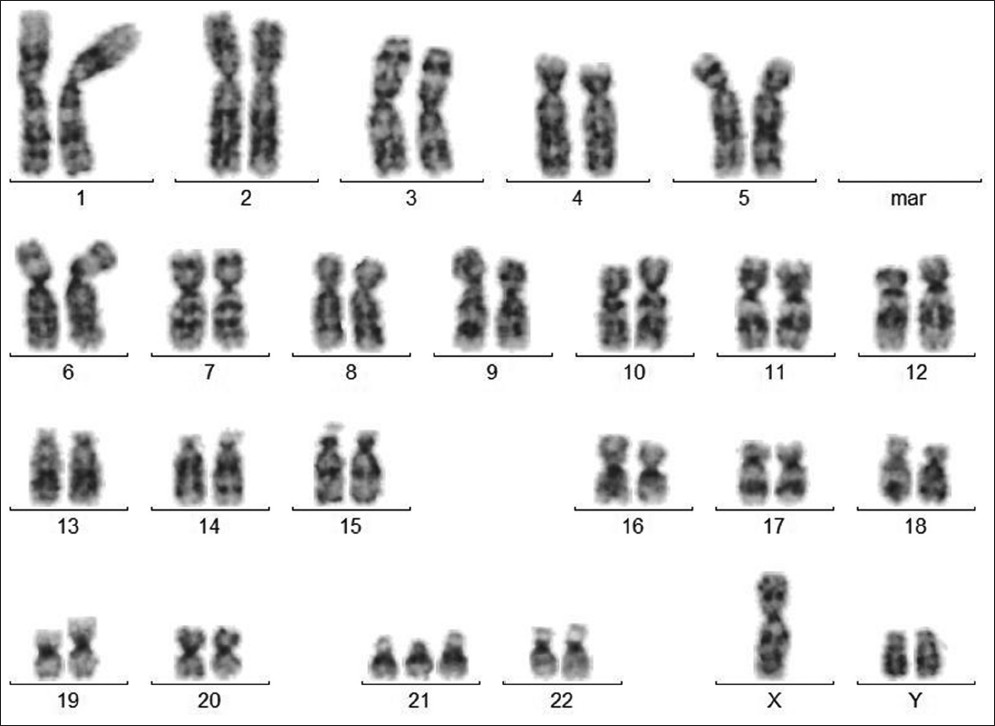 |
Mosaic double aneuploidy: Down syndrome and XYY |
p. 346 |
Mayur Parihar, Beena Koshy, Vivi Miriam Srivastava
DOI:10.4103/0971-6866.120825 PMID:24339550Chromosomal abnormalities are seen in nearly 1% of live born infants. We report a 5-year-old boy with the clinical features of Down syndrome, which is the most common human aneuploidy. Cytogenetic analysis showed a mosaicism for a double aneuploidy, Down syndrome and XYY. The karyotype was 47, XY,+21[19]/48, XYY,+21[6]. ish XYY (DXZ1 × 1, DYZ1 × 2). Mosaic double aneuploidies are very rare and features of only one of the aneuploidies may predominate in childhood. Cytogenetic analysis is recommended even if the typical features of a recognized aneuploidy are present so that any associated abnormality may be detected. This will enable early intervention to provide the adequate supportive care and management. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
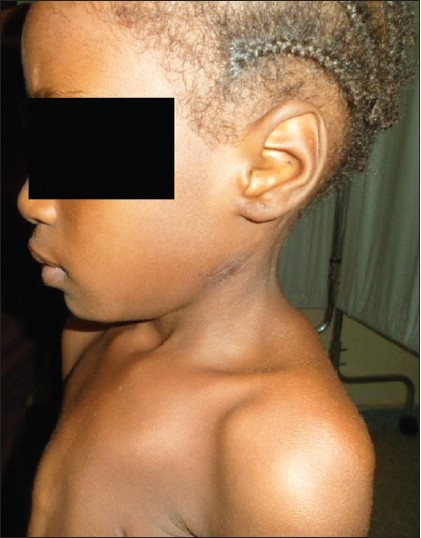 |
Poland syndrome a rare congenital anomaly |
p. 349 |
Aliyu Ibrahim, Abdallah Ramatu, Akhiwu Helen
DOI:10.4103/0971-6866.120824 PMID:24339551Poland syndrome is a rare congenital anomaly classically consisting of unilateral hypoplasia of the sternocostal head of the pectoralis major muscle and ipsilateral brachysyndactyly. It was first described by Alfred Poland in 1840 and may occur with different gravity. Our patient is an eight-year-old Nigerian girl with left-sided anterior chest wall defect with no detectable structural heart abnormality but presented with repeated episodes of syncopal attacks following minor trauma to the anterior chest wall. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
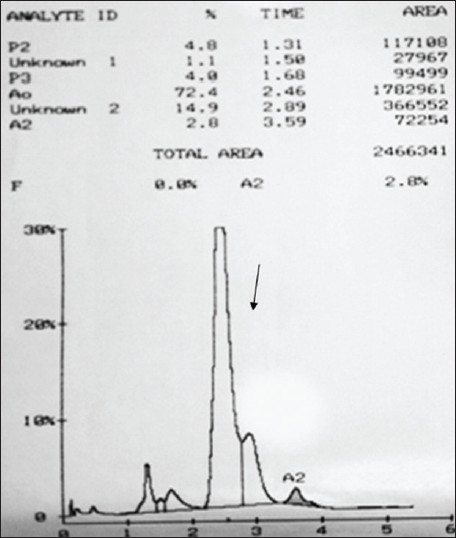 |
Hemoglobin Fontainebleau [a21(B2)Ala>Pro]: The second report from India |
p. 352 |
Ranjeet Singh Mashon, Sona Nair, Pratibha Sawant, Roshan B Colah, Kanjaksha Ghosh, Sheila Das
DOI:10.4103/0971-6866.120822 PMID:24339552Structural hemoglobin (Hb) variants are mainly due to point mutations in the globin genes resulting in single amino acid substitutions. Until date, about 200 alpha chain variants have been identified and they are usually detected during the hemoglobinopathy screening programs. Under a community control program for hemoglobinopathies, which involved screening of antenatal cases followed by prenatal diagnosis if indicated. Here, we report a rare alpha globin gene variant Hb Fontainebleau [a21(B2)Ala>Pro] detected in the heterozygous condition in a 35-year-old pregnant lady screened during this program. This is the second report of this alpha globin variant from India. Unlike the earlier case from India where Hb Fontainebleau was reported in a neonate who was also a carrier of Hb Sickle and had no clinical problems, this case presented with a bad obstetric history associated with the secondary infertility. However, the presence of the variant and the obstetric complications may be unrelated. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
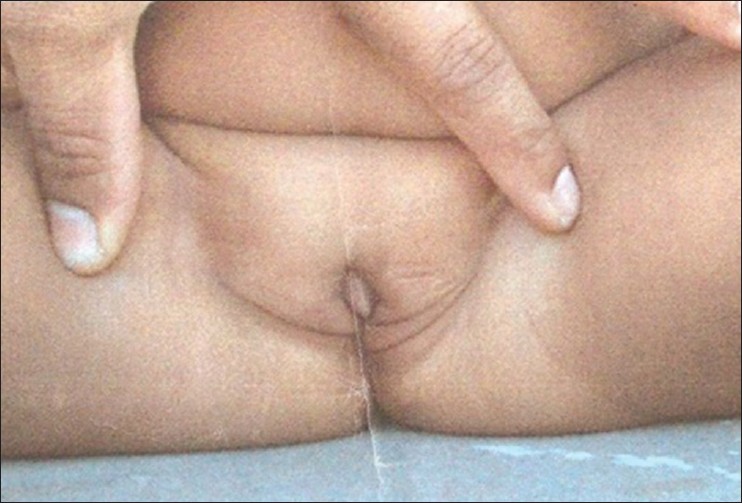 |
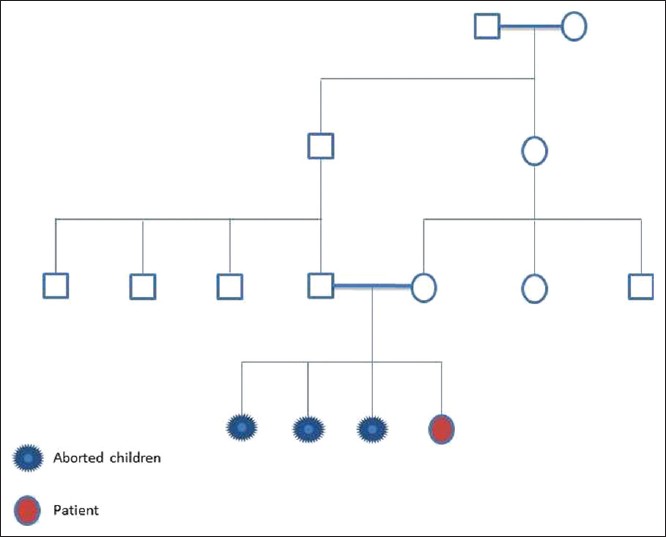
Genetic analysis of a family with complete androgen insensitivity syndrome  |
p. 355 |
Sunil Kumar Kota, Kotni Gayatri, Siva Krishna Kota, Sruti Jammula
DOI:10.4103/0971-6866.120820 PMID:24339553Androgen insensitivity causes impaired embryonic sex differentiation leading to developmental failure of normal male external genitalia in 46 XY genetic men. It results from diminished or absent biological actions of androgens, which is mediated by the androgen receptor (AR) in both the embryo and secondary sexual development. Mutations in the AR located on the X chromosome are responsible for the disease. Almost 70% of affected individuals inherit the mutation from their carrier mother. We hereby report a 10-year-old girl with all the characteristics of complete androgen insensitivity syndrome (CAIS). Similar scenario was observed in 3 maternal aunts, Sequencing of the AR gene in all the family members revealed C 2754 to T transition in exon 6. It was concluded that the C 2754 to T transition rendered the AR incapable of both ligand-binding and activating the transcription and was the cause of CAIS in the patient. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
Type 2 diabetes mellitus: An unusual association with Down's syndrome |
p. 358 |
Sunil Kumar Kota, Prabhas Ranjan Tripathy, Siva Krishna Kota, Sruti Jammula
DOI:10.4103/0971-6866.120818 PMID:24339554Down's syndrome (DS) is known to be associated with autoimmune disease including type 1 diabetes. To the best of our knowledge, there are no reports of DS with type 2 diabetes mellitus in the literature. We hereby report two cases of DS with type 2 diabetes. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
A case of primary amenorrhea with 46+XY genotype from Kashmir Valley |
p. 360 |
Shahid Mudassir Baba, Aga Syed Sameer, Mushtaq A Siddiqi
DOI:10.4103/0971-6866.120816 PMID:24339555Primary amenorrhea is one of the common reproductive disorder affecting females. It leads to the absence of menarche in the reproductive age group in females and/or complete absence of reproductive organs. There are many causes which lead to PA, including genetic aberrations which are the leading factors. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Hypoparathyroidism-retardation-dysmorphism syndrome |
p. 363 |
Kalenahalli Jagadish Kumar, Halasahalli Chowdegowda Krishna Kumar, Vadambal Gopalakrishna Manjunath, Sangaraju Mamatha
DOI:10.4103/0971-6866.120814 PMID:24339556Congenital hypoparathyroidism, growth retardation and facial dysmorphism is a rare autosomal recessive disorder seen among children born to consanguineous couple of Arab ethnicity. This syndrome is commonly known as Sanjad-Sakati or hypoparathyroidism-retardation-dysmorphism syndrome (HRD). We report 13-year-old Hindu boy with hypoparathyroidism, tetany, facial dysmorphism and developmental delay, compatible with HRD syndrome. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
A novel ABCB11 mutation in an Iranian girl with progressive familial intrahepatic cholestasis |
p. 366 |
Sassan Saber, Reza Vazifehmand, Iman Bagherizadeh, Mahbubeh Kasiri
DOI:10.4103/0971-6866.120813 PMID:24339557Progressive familial intrahepatic cholestasis is an autosomal recessive liver disorder caused by (biallelic) mutations in the ATP8B1 of ABCB11 gene. A nine-year-old girl with cholestasis was referred for genetic counseling. She had a family history of cholestasis in two previous expired siblings. Genetic analysis of the ABCB11 gene led to the identification of a novel homozygous mutation in exon 25. The mutation 3593- A > G lead to a missense mutation at the amino acid level (His1198Arg). This mutation caused PFIC2 due to abnormal function in the bile salt export pump protein (BSEP). |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
 |
Genetic mutations in Gorlin-Goltz syndrome |
p. 369 |
Muthumula Daneswari, Mutjumula Swamy Ranga Reddy
DOI:10.4103/0971-6866.120810 PMID:24339558Gorlin-Goltz syndrome is a rare multisystemic disease inherited in a dominant autosomal at a high level of penetrance and variable expressiveness. It is mainly characterized by basal cell carcinoma, odontogenic keratocyst and skeletal anomalies. Diagnosis is based upon established major and minor clinical and radiographic criteria and gene mutation analysis. This article presents a case of Gorlin-Goltz syndrome, its genetic predisposition, diagnosis and management. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
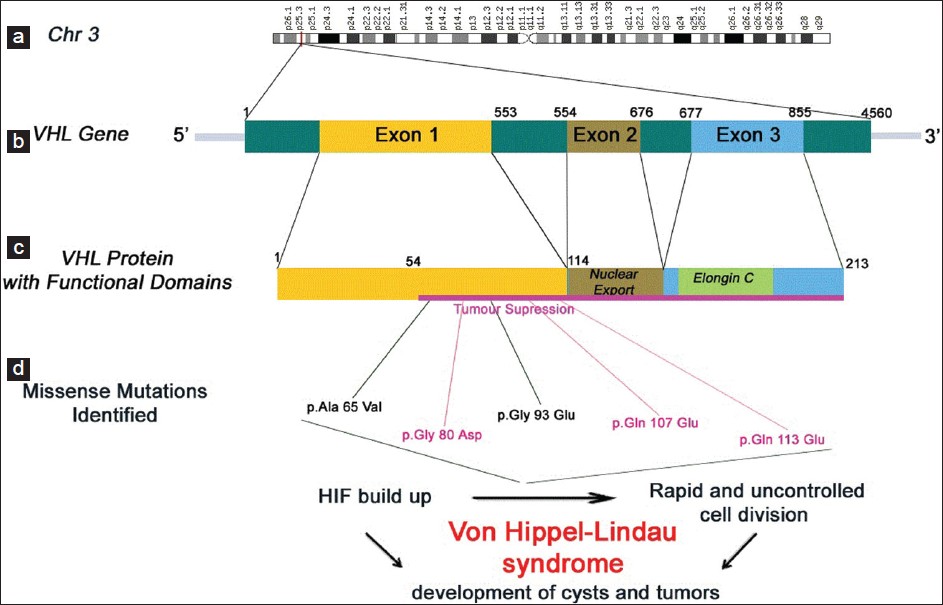 |
Novel three missense mutations observed in Von Hippel-Lindau gene in a patient reported with renal cell carcinoma |
p. 373 |
Pasupuleti Santhosh Kumar, Katari Venkatesh, Lokanathan Srikanth, Potukuchi Venkata Gurunadha Krishna Sarma, Akkamgari Ramprasad Reddy, Srinivasan Subramanian, Bobbidi Venkata Phaneendra
DOI:10.4103/0971-6866.120809 PMID:24339559Von Hippel-Lindau (VHL) disease is an autosomal dominant hereditary cancer syndrome that predisposes to the development of a variety of benign and malignant tumors, especially cerebellar hemangioblastomas, retinal angiomas and clear-cell renal cell carcinomas (RCC). We have identified of VHL gene using immunohistochemistry in a patient who was diagnosed for RCC. In order to understand the involvement of mutation in the VHL gene exon 1 was amplified and sequenced (accession number: JX 401534). The sequence analysis revealed the presence of novel missense mutations c.194 C>T, c.239 G>A, c.278 G>A, c.319 C>G, c. 337 C > G leading to the following variations p.Ala 65 Val, p.Gly 80 Asp, p.Gly 93 Glu, p.Gln 107 Glu, p.Gln 113 Glu in the protein. |
| [ABSTRACT] [HTML Full text] [PDF] [Mobile Full text] [EPub] [Citations (2) ] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|
|
| LETTER TO THE EDITOR |
|
|
|
|
Possible impact of factor V Leiden genotype on warfarin induced bleeding |
p. 377 |
Tejasvita Gaikwad, Kanjaksha Ghosh, Shrimati Shetty
DOI:10.4103/0971-6866.120807 PMID:24339560 |
| [HTML Full text] [PDF] [Mobile Full text] [EPub] [PubMed] [Sword Plugin for Repository]Beta |
|
|
|
|
|