|
|
Levosimendan vs Dobutamine chez des patients présentant une insuffisance cardiaque en décompensation aiguëL'essai clinique randomisé SURVIVE
Alexandre Mebazaa, MD, PhD;
Markku S. Nieminen, MD, PhD;
Milton Packer, MD;
Alain Cohen-Solal, MD, PhD;
Franz X. Kleber, MD;
Stuart J. Pocock, PhD;
Roopal Thakkar, MD;
Robert J. Padley, MD;
Pentti Põder, MD, PhD;
Matti Kivikko, MD, PhD; Pour les investigateurs SURVIVE
RÉSUMÉ
| |
Contexte Parce que l'insuffisance cardiaque en décompensation
aiguë est à l'origine d'une morbidité et d'une
mortalité importante, il y a un besoin en agents thérapeutiques
qui, tout au moins, amélioreraient l'état hémodynamique
et soulageraient les symptômes, sans conséquences
indésirables sur la survie.
Objectifs Evaluer l'effet d'une perfusion intraveineuse à
court terme de levosimendan ou de dobutamine sur la survie à long
terme.
Schéma, environnement et patients L'étude SURVIVE (la
survie des patients présentant une insuffisance cardiaque en
décompensation aiguë et nécessitant un support inotropique
intraveineux) est un essai randomisé en double aveugle, comparant
l'efficacité et la sécurité de l 'administration
intraveineuse de levosimendan ou de dobutamine chez 1327 patients,
hospitalisés pour une insuffisance cardiaque en décompensation
aiguë ayant nécessité un support inotropique. L'essai avait
été mené dans 75 centres répartis dans 9 pays avec
randomisation des patients entre mai 2003 et décembre 2004.
Traitements Administration intraveineuse de levosimendan (n=664) ou
de dobutamine (n=663)
Principal critère de jugement Mortalité toutes causes
confondues à 180 jours
Résultats Le décès, toutes causes confondues,
s'était produit chez 173 patients (26%) appartenant au groupe
levosimendan et 185 patients (28%) du groupe dobutamine (risque relatif, 0.91,
intervalle de confiance 95%, 0.74-1.13, P=.40). Par rapport au groupe
dobutamine, le groupe levosimendan avait connu une diminution plus importante
du taux de peptide natriurétique de type B à 24 heures et
maintenue durant 5 jours (P<0.01 pour tous les points d'observation).
Aucune différence statistique n'était observée pour les
critères d'évaluation secondaires (mortalité toutes
causes confondues à 31 jours, nombre de jours passés en vie et
non hospitalisé, évaluation globale du patient,
évaluation de dyspnée après 24h et la mortalité
cardiovasculaire à 180 jours). L'insuffisance cardiaque était
plus fréquente dans le groupe dobutamine. La fibrillation auriculaire,
l'hypocalémie et la migraine étaient plus fréquentes dans
le groupe levosimendan.
Conclusion Malgré une diminution initiale du taux plasmatique
de peptide natriurétique de type B chez les patients du groupe
levosimendan comparativement au groupe dobutamine, le levosimendan n'apportait
aucune baisse significative de la mortalité toutes causes confondues et
n'affectait aucuns des résultats cliniques secondaires.
Trial Registration
clinicaltrials.gov
Identifier: NCT00348504
JAMA.
2007;297:1883-1891
L'insuffisance cardiaque en décompensation aiguë (ADHF,
Acute Decompensated Heart Failure) demeure une cause fréquente
d'hospitalisation dans le monde mais il n'est pas clairement défini
comment les patients, admis à la suite d'une
détérioration clinique, doivent être pris en charge. Les
patients sont habituellement traités avec des diurétiques et des
vasodilatateurs; ceux qui présentent des signes probants
d'hypoperfusion périphérique pouvant également recevoir
des agents inotropes positifs, généralement de la dobutamine ou
de la milrinone. Ces agents inotropes positifs améliorent l'état
hémodynamique et les symptômes en augmentant le niveau d'AMP
cyclique intracellulaire dans le coeur défaillant mais ont
également été associés à une
élévation du risque de décès et d'autres accidents
cardiovasculaires.1,2
Le levosimendan est un agent pharmacologique qui exerce des effets
inotropiques positifs par fixation calcium-dépendante sur la troponine
C cardiaque, sensibilisant les myofilaments au
calcium.3,4
Il possède également des propriétés
vasodilatatrices par son action favorisant l'ouverture ATP-dépendante
des canaux
potassiques5
et par ses effets
anti-ischémiques6.
Les études cliniques ont montré que le levosimendan accroissait
le débit cardiaque et abaissait les pressions de remplissage cardiaque
et était également associé à une réduction
des symptômes cardiaques, du risque de décès et des
hospitalisations7,9.
Contrairement aux autres agents inotropes positifs, ses actions principales
n'impliquent pas d'interactions avec les récepteurs
β-adrénergiques10,11.
Sa comparaison avec l'agoniste des récepteurs
β-adrénergiques, la dobutamine, dans l'essai
LIDO12
(Perfusion de Levosimendan vs Dobutamine) avait montré des effets
hémodynamiques supérieurs et une association à un risque
de décès, à 31 jours et 180 jours, plus faible dans les
analyses secondaires et post-hoc.
|
|
|
|
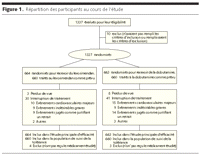
Figure 1.. Répartition des participants au cours de l'étude
|
|
|
SURVIVE (Survie de patients présentant une insuffisance cardiaque
aiguë nécessitant un support inotropique intraveineux) a
été, à notre connaissance, le premier essai sur la survie
réalisé chez des patients présentant une ADHF. L'essai
SURVIVE évaluait l'effet d'une perfusion intraveineuse de levosimendan
ou de dobutamine à court terme sur la survie à long terme.
METHODES
L'essai SURVIVE a été conduit dans 75 centres en Autriche,
Finlande, France, Allemagne, Israël, Lituanie, Pologne, Russie et le
Royaume-Uni. Le protocole d'étude avait été
approuvé par des commissions d'éthiques indépendantes et
réalisé conformément à la déclaration
d'Helsinki13
ainsi que des règlements en vigueurs. L'essai avait été
conçu, mis en place, réalisé et supervisé par
l'organisme financeur et le comité de pilotage. Un conseil de
surveillance et de sécurité indépendant avait libre
accès aux résultats et évaluait périodiquement les
résultats portant sur la sécurité.
Patients étudiés
L'étude incluait des patients de 18 ans ou plus, hospitalisés
pour une ADHF et ayant donnés leur consentement éclairé
par écrit et signé. Tous les patients avaient une fraction
d'éjection de 30% ou inférieure au cours des 12 derniers mois et
requéraient un support inotropique intraveineux, ce que
démontraient la réponse insuffisante aux diurétiques
intraveineux et/ou aux vasodilatateurs et au moins l'un des résultats
suivants lors du dépistage: (1) dyspnée au repos ou ventilation
mécanique pour ADHF, (2) oligurie d'origine non hypovolémique,
ou (3) une pression pulmonaire capillaire de 18 mm Hg ou supérieure
et/ou un index cardiaque de 2.2L/min/m2 ou inférieur.
Les critères d'exclusion incluaient une obstruction
sévère de l'écoulement ventriculaire, une pression
sanguine systolique systématiquement inférieure à 85 mm
Hg ou un rythme cardiaque systématiquement à 130/min ou plus,
l'administration d'inotropes intraveineux durant l'hospitalisation index (sauf
dopamine 2 µg/kg/min ou digitaline), des antécédents de
torsades de pointes, un taux de créatinine sérique
supérieur à 5.1 mg/dL (450 µmol/L) ou une dialyse.
Plan d'étude
SURVIVE est une étude randomisée, multicentrique, en double
aveugle et en groupes parallèles. La randomisation avait
été réalisée en deux étapes
(FIGURE 1). Dans un premier
temps, des flacons contenant les médicaments à étudier
étaient numérotés aléatoirement utilisant un
système de blocs permutés. Les patients étaient ensuite
randomisés au niveau central, employant un système vocal
interactif qui assurait une répartition 1:1 entre le levosimendan et la
dobutamine. La randomisation était stratifiée en employant un
algorithme de tirage au sort biaisé avec l'existence
d'antécédent d'ADHF et le pays d'origine comme facteurs.
Lors de la période de traitement, les patients ont été
randomisés pour recevoir deux perfusions intraveineuses en double
aveugle. Du levosimendan et un placebo de la dobutamine dans le groupe
levosimendan ou de la dobutamine et un placebo du levosimendan dans le groupe
dobutamine. Une dose de charge de levosimendan (12 µg/kg) ou de placebo du
levosimendan était administrée sur 10 minutes, suivie d'une
perfusion (0.1 µg/kg/minute) de 50 minutes. Le taux était
augmenté de 0.2 µg/kg/minute sur 23 heures supplémentaires ou
jusqu'à la dose tolérée. La perfusion de la dobutamine ou
du placebo de la dobutamine était initiée à un taux de 5
µg/kg/minute pouvant être accru à la discrétion de
l'investigateur, jusqu'à un taux de 40 µg/kg/minute. La perfusion
était maintenue aussi longtemps que cliniquement nécessaire
(avec un minimum de 24 heures) et ajustée en fonction de la situation
clinique de chaque patient.
Le niveau plasmatique de peptide natriurétique de type B
était mesuré au bout de 1, 3 et 5 jours. Le décès
ou l'hospitalisation était noté sur la période de 180
jours. Si les patients nécessitaient un support inotropique additionnel
au cours de la période d'étude, l'intention était de
maintenir l'insu en ré-administrant le médicament initialement
attribué avec la dose thérapeutique initiale. Cependant, ceci
n'était pas un impératif et son non suivi n'était pas
considéré comme une violation du protocole. Si la
ré-administration se produisait dans les sept jours suivant la
perfusion initiale, le levosimendan était administré sans dose
de charge, à 0.1 µg/kg/minute.
|
|
|
|
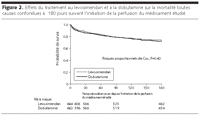
Figure 2.. Effets du traitement au levosimendan et à la dobutamine sur la
mortalité toutes causes confondues à 180 jours suivant
l'initiation de la perfusion du médicament étudié.
|
|
|
Critères d'évaluation
Le critère d'évaluation principal était la
mortalité toutes causes confondues au cours des 180 jours suivant la
randomisation. Les critères secondaires incluaient la mortalité
toutes causes confondues sur 31 jours, la variation du niveau de BNP sur 24
heures, comparé à la valeur initiale, le nombre de jours
passés en vie et non hospitalisé au cours des 180 jours, les
variations dans l'auto-évaluation d'une dyspnée par le patient
à 24 heures, auto-évaluation globale fait par le patient
à 24 heures, la mortalité cardiovasculaire sur les 180
jours.
Pour surveiller la sécurité, les évènements
indésirables étaient recueillis pendant les 31 jours suivant
l'administration initiale du médicament étudié et durant
toutes les ré-administrations en aveugle.
Analyses statistiques
La taille de l'échantillon était déterminée par
l'objectif d'évaluer les différences de mortalité, toutes
causes confondues, entre le groupe levosimendan et dobutamine à 180
jours. L'essai a été conduit jusqu'à ce que 330
décès se soient produits, fournissant une puissance de 85%
(?=0.05) pour détecter une réduction relative du risque de
décès de 25% entre les groupes de traitements, en supposant que
la mortalité à 180 jours était à 25.3%. L'objectif
initial en nombre de patients, se basant sur l'étude LIDO, était
de 700 mais suite à une évaluation en aveugle de la
mortalité après 131 décès, avait été
élargi à 1320 pour atteindre le nombre de 330
décès recherché.
Afin de contrôler l'erreur de type I résultant de deux
analyses intérimaires, la limite d'arrêt de Haybittle-Peto avait
été utilisée; les différences de risque entre les
deux traitements, au terme de l'essai, étaient
considérées comme significatives à une valeur de P
inférieure à 0.05. L'analyse de survie, en intention de
traitement, était basée sur la randomisation des patients. Les
courbes de survie cumulées étaient tracées en tant que
temps jusqu'à l'évènement, en utilisant la méthode
de Kaplan-Meier; la significativité des différences était
évaluée avec le model de régression à risques
proportionnels de Cox, en prenant le traitement comme unique covariable. Le
model de Cox était également utilisé pour analyser des
interactions potentielles dans les combinaisons sous-groupe/traitement X,
utilisant le traitement, le sous-groupe, et les combinaisons
sous-groupe/traitement X comme covariables. Les analyses
prédéfinies pour les sous-groupes portaient sur les valeurs
initiales des variables suivantes: le sexe, l'âge, les
antécédents d'ADHF, infarctus aigu du myocarde à la
première hospitalisation, le niveau de créatinine
sérique, l'oligurie, l'utilisation de β-bloquants, utilisation
d'inhibiteur de l'enzyme de conversion de l'angiotensine ou d'un antagoniste
des récepteurs de l'angiotensine II, la pression sanguine systolique,
le rythme cardiaque, la présence de dyspnée au repos et la
ventilation mécanique pour ADHF. Le recours au model de Cox dans les
sous-groupes était considéré davantage si la valeur P de
l'interaction était de 0.10 ou inférieure.
|
|
|
|
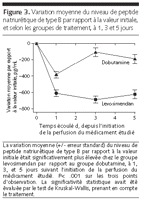
Figure 3.. Variation moyenne du niveau de peptide natriurétique de type B par
rapport à la valeur initiale, et selon les groupes de traitement,
à 1, 3 et 5 jours
|
|
|
La comparaison des variables qualitatives comme l'évaluation de la
dyspnée, l'évaluation globale du patient et le nombre de jours
passés en vie sans hospitalisation, fait appel au test de
Cochran-Mantel-Haenszel, en considérant uniquement le traitement. Les
variations du niveau en BNP étaient analysées avec le test de
Kruskal-Wallis. La comparaison du taux d'incidence des
évènements indésirables entre les différents
groupes était réalisée avec le test exact de Fisher et
pour les autres variables, la variation moyenne par rapport aux valeurs
initiales était établie par analyse de la covariance avec les
valeurs initiales comme covariables. Les différences entre les
traitements, sur analyse de la variation moyenne par rapport aux valeurs
initiales en électrocardiogramme et à l'observation des signes
vitaux avaient été testées par l'analyse de la covariance
avec les valeurs initiales comme covariables. Les analyses statistiques ont
été réalisées avec SAS version 8.2 (SAS Institute
Inc, Cary, NC) et la significativité était constatée
à une valeur P de 0.05 ou inférieure.
RESULTATS
Population étudiée
Au total, 1327 patients hospitalisés pour une ADHF avaient
été randomisés, entre mars 2003 et décembre 2004,
entre le groupe levosimendan (n=664) et le groupe dobutamine (n=663). Sur
cette population définie par l'intention de traitement, 1320 patients
(660 dans chaque groupe) recevant le médicament étudié
avaient été inclus dans la population de suivi de la
tolérance (Figure
1).
Les patients, randomisés au levosimendan ou à la dobutamine,
étaient similaires au regard des aspects prétraitements
(TABLE 1) et des
médicaments concomitants. Ils avaient un niveau de BNP
élevé et la grande majorité (n=1171, 88%) avaient un
antécédent d'ADHF.
|
|
|
|
Tableau 1.. Données démographiques et caractéristiques initiales
des patients*
|
|
|
Au delà de la première heure incluant la dose de charge, le
levosimendan était perfusé en continu, avec un taux moyen de 0.2
(ET=0.02) µg/kg/minute pendant 23.4 (ET=2.9) heures; la dobutamine à
un taux de 5.9 (ET=2.6) µg/kg/minute pendant 39.3 (ET=44.4) heures. Sur la
période de 180 jours, 102 (<8%) patients (48 du groupe levosimendan
et 54 du groupe dobutamine) avaient reçu en insu une reperfusion du
médicament étudié. 75 (11%) patients du groupe
levosimendan avaient également reçu, pendant l'étude et
de manière ouverte, de la dobutamine (n=72) ou du levosimendan (n=3)
tandis que 79 (12%) patients du groupe dobutamine avaient reçu, de
manière ouverte, de la dobutamine (n=74) ou du levosimendan (n=5).
Critères principal de jugement et critères secondaires
Sur les 180 jours suivant la perfusion du médicament
étudié, il s'était produit 173 décès (26%)
dans le groupe levosimendan, et 185 décès dans le groupe
dobutamine (28%) (Risque relatif 0.91 [IC 95%, 0.74-1.13] P=0.40;
FIGURE 2). L'observation de la
mortalité, toutes causes confondues, à 31 jours et de la
mortalité cardiovasculaire à 180 jours n'avaient montré
aucune différence entre les deux groupes de traitement
(TABLE 2).
|
|
|
|
Tableau 2.. Critère d'évaluation principale, secondaires et
mortalité toutes causes confondues post hoc.*
|
|
|
La diminution du BNP plasmatique était plus importante chez le
groupe levosimendan à 24 heures, 3 jours et 5 jours (P<0.001 pour
tous les cas) (FIGURE 3) Les
autres variables étaient identiques entre les deux groupes de
traitement (TABLE 2).
Des analyses de sous-groupes avaient été
réalisées pour évaluer l'influence de
caractéristiques initiales préétablis sur la
différence de survie entre les patients des deux groupes
(TABLE 3 et
TABLE 4). La plupart des
analyses n'avaient montré aucune corrélation. Cependant, un
antécédent d'insuffisance cardiaque influençait la
différence entre les deux groupes à 31 jours (corrélation
entre traitement X et antécédent d'insuffisance cardiaque,
P=0.05) mais pas à 180 jours. Chez 88% des patients avec un
antécédent d'insuffisance cardiaque, le risque de
décès à 31 jours tendait à diminuer chez les
patients du groupe levosimendan comparativement au groupe dobutamine.
Inversement, dans le sous-groupe exempt d'antécédents
d'insuffisance cardiaque (12%), on assistait à une augmentation
numérique du décès dans le groupe levosimendan.
|
|
|
|
Tableau 3.. Influence des valeurs et caractéristiques initiales sur la survie
à 31 jours*
|
|
|
|
|
|
|
Tableau 4.. Influence des valeurs et caractéristiques initiales sur la survie
à 180 jours*
|
|
|
Sécurité et tolérance
Les diminutions de la pression sanguine systolique et diastolique
étaient initialement majorées dans le groupe levosimendan par
rapport au groupe dobutamine. Ces différences persistaient après
l'interruption de la perfusion des médicaments étudiés
(FIGURE 4). L'augmentation du
rythme cardiaque était majorée dans le groupe levosimendan par
rapport au groupe dobutamine (FIGURE
4) et se maintenait pendant 5 jours.
|
|
|
|
Figure 4.. Variations moyennes de la pression sanguine systolique, de la pression
sanguine diastolique et du rythme cardiaque sur 5 jours, par rapports aux
valeurs initiales, et selon le groupe de traitement.
|
|
|
Comparés aux patients traités à la dobutamine, les
patients traités au levosimendan étaient moins susceptibles de
connaître une insuffisance cardiaque (P=0.02) et plus susceptibles de
connaître une fibrillation auriculaire (P=0.05), une hypocalémie,
(P=0.02) et des migraines (P=0.01) durant les 31 premiers jours suivant
l'administration du médicament étudié
(TABLE 5). Les groupes de
traitement étaient similaires au regard de la fréquence
d'hypotension, d'insuffisance rénale, des arythmies ventriculaires ou
des antécédents de torsades de pointes. L'intervalle QT
n'augmentait pas avec le levosimendan et ne montrait pas de différence
entre les deux groupes.
|
|
|
|
Tableau 5.. Évènements indésirables dus aux
traitements*
|
|
|
COMMENTAIRES
Les patients hospitalisés pour une ADHF présentent un risque
élevé de décès et de ré-hospitalisation
dans les mois suivant l'admission.
14,15
Les preuves existantes suggèrent que le risque à court terme
peut être influencé par le traitement16, plusieurs agents
thérapeutiques dont la dobutamine, la milrinone et la nésiritide
ayant été associés à un accroissement
précoce du risque de
décès.1,17,18
Dans cette étude, nous avons comparé le levosimendan, qui agit
en sensibilisant la réponse des myofilaments cardiaques au calcium et
en facilitant l'ouverture ATP-dépendante des canaux potassiques,
affectant peu l'AMP cyclique intracellulaire, à la dobutamine, l'agent
thérapeutique le plus utilisé dans le traitement de l'ADHF, et
qui agit principalement en augmentant la quantité d'AMP cyclique
intracellulaire. Dans l'essai
LIDO,12 les
patients affectés au groupe levosimendan présentaient un risque
de décès moindre que ceux affectés au groupe dobutamine
mais ce bénéfice n'était pas le critère
d'évaluation principal.
L'étude SURVIVE est le premier essai prospectif, randomisé
à suivre la survie à long terme des patients présentant
une ADHF. Les résultats n'avaient montré aucune
différence significative entre le levosimendan et la dobutamine sur la
mortalité toutes causes confondues à 31 jours et 180 jours
suivant la perfusion du médicament étudié.
L'étude avait mis en évidence une baisse notable du niveau de
BNP chez les patients traités au levosimendan par rapport à ceux
traités à la dobutamine, sur 5 jours. Il y a une
cohérence entre la pharmacocinétique des deux médicaments
et les différences d'effets observées; le métabolite
actif du levosimendan atteint sont pic aux alentours de 3 jours suivant la
perfusion et possède une demi-vie de 80
heures.19
Contrairement à la dobutamine qui a une demivie plus courte et aucun
métabolite connu. De ce fait, il faut souligner que dans cet essai, les
différences numériques sur le taux de survie entre les deux
médicaments avaient été observées très
tôt, faisant immédiatement suite à l'interruption du
traitement (risque relatif 0.72 [IC 95%, 0.44-1.16] sur l'analyse 5 jours
post-hoc) mais ces différences de risques relatifs s'étaient
résorbées en l'absence d'un traitement continu sur le suivi
à long terme.
Une différence qui ne s'appliquerait qu'à un sous-groupe de
patients présentant une ADHF pourrait également expliquer
l'absence de différence sur la mortalité entre les deux groupes
de traitement. Dans SURVIVE, la randomisation était stratifiée
par l'antécédent d'insuffisance cardiaque, se basant sur des
signes probants, observés préalablement, que celui-ci pourrait
affecter la mortalité due à
l'ADHF20 et
les différences entre les traitements. De ce fait, il est
intéressant de noter qu'un antécédent d'insuffisance
cardiaque influençait les différences que nous avions
observées. Plus précisément, les différences en
faveur du levosimendan, sur 31 jours, étaient plus évidentes
chez des patients ayant déjà un antécédent
d'insuffisance cardiaque que chez ceux où l'installation de
l'insuffisance cardiaque est récente, probablement à cause de
l'utilisation importante de β-bloquants chez les patients affectés
d'une insuffisance cardiaque chronique. Les antagonistes du récepteur
β pourraient interférer avec les bénéfices
hémodynamiques apportés par la
dobutamine21,22
ou potentialiser les actions du
levosimendan12,23
sur la circulation ou les deux à la fois.
Une troisième explication viendrait de la différente
stratégie de dosage employée pour la dobutamine dans
l'étude SURVIVE, qui empêcherait de reproduire la
mortalité sur 180 jours, observée dans l'étude
LIDO,12 une
étude hémodynamique où tous les patients avaient subis un
cathétérisme artériel pulmonaire. L'approche
élaborée dans SURVIVE ne correspondait pas à la
méthode de dosage de la dobutamine appliquée dans LIDO dans
laquelle les patients du groupe dobutamine avaient reçu en dose
initiale 5 µg/kg/minute avec une augmentation de la dose à 10
µg/kg/minute si les objectifs hémodynamiques n'étaient pas
atteints. Dans SURVIVE, la dose de dobutamine pouvait être
augmentée jusqu'à 40 µg/kg/minute pour atteindre les
objectifs cliniques, cependant elle avait été administrée
à la dose relativement faible de 6 µg/kg/minutes sur une moyenne de
39 heures. Le régime de dosage de la dobutamine était
déterminé par le médecin traitant en insu et selon les
besoins du patient. Par ailleurs, la perfusion de la dobutamine avait
été interrompue au bout de 24 heures dans l'étude LIDO.
La stratégie de dosage individualisée adoptée pour la
dobutamine dans l'étude SURVIVE pourrait être à l'origine
de la mortalité de 28% à 180 jours, comparée au 38%
observée dans l'étude LIDO. Par contraste, le levosimendan
était administré à une dose fixe dans l'étude
SURVIVE et LIDO, avec le même taux de mortalité sur 180 jours,
égale à 26%. Par conséquent, les futurs essais devraient
permettre également l'individualisation de la dose de levosimendan.
Par rapport aux patients traités à la dobutamine, les
patients traités au levosimendan étaient plus susceptibles de
connaître une baisse initiale de la pression systolique et la pression
diastolique sanguines (Figure
4). Cette dernière, probablement associée à
la mode d'administration du levosimendan et surtout à la dose
administrée en bolus, pourrait être responsable de la survenue de
fibrillation auriculaire et d'autres effets préjudiciables comme le
décès. Par conséquent, les futures études
devraient analyser la façon optimale d'administrer le levosimendan.
L'hypocalémie était systématiquement observée
dans SURVIVE, comme dans les autres
essais.7,12
Les mécanismes de l'hypocalémie restent à
élucider.
En conclusion, l'essai SURVIVE n'avait démontré aucune
différence sur la survie entre le levosimendan et la dobutamine dans un
suivi à long terme malgré la réduction précoce du
niveau plasmatique de BNP avec le levosimendan. Ces résultats
pourraient être corrélés à la courte durée
du traitement, l'effectivité sélective du levosimendan pour des
sous-groupes spécifiques, ou un manque réel de différence
entre les deux médicaments. Des études plus poussées sont
nécessaires pour faire la distinction entre ces différentes
possibilités.
Informations sur les auteurs
Correspondance: Alexandre Mebazaa, MD, PhD, Hôpital
Lariboisière, 2 Rue Ambroise Paré, Paris, France 75475 Cedex 10
(alexandre.mebazaa{at}lrb.aphp.fr).
Contributions des auteurs: Les Drs Mebazaa et Nieminen ont eu un
accès complet à toutes les données de l'étude et
acceptent la responsabilité de l'intégrité des
données et de l'exactitude de l'analyse des données.
Concept et schéma de l'étude: Mebazaa, Nieminen,
Packer, Cohen-Solal, Kleber, Pocock, Pöder, Kivikko.
Recueil des données: Nieminen, Padley, Pöder,
Kivikko.
Analyse et interprétation des données: Mebazaa,
Nieminen, Packer, Cohen-Solal, Kleber, Pocock, Thakkar, Padley,
Pöder.
Rédaction du manuscrit: Mebazaa, Nieminen, Packer,
Cohen-Solal, Kleber, Pocock, Thakkar, Padley.
Revue critique du manuscrit: Mebazaa, Nieminen, Packer,
Cohen-Solal, Kleber, Pocock, Thakkar, Padley, Pöder, Kivikko.
Analyse statistique: Pocock, Padley.
Obtention du financement: Padley, Pöder.
Aide administrative, technique ou matérielle: Thakkar,
Padley, Pöder, Kivikko.
Supervision de l'étude: Mebazaa, Nieminen, Packer,
Cohen-Solal, Kleber, Thakkar, Padley.
Liens financiers: Le Dr Mebazaa a déclaré être
consultant chez Abbott, Orion Pharma, Protein Design Biopharma, et Sigma-Tau
et avoir reçu des honoraires d'Abbott, Guidant, et Edwards Life
Sciences. Le Dr Nieminen a déclaré être consultant chez
Abbott, Orion Pharma, Scios, Medtronic, et Pfizer. Le Dr Cohen-Solal a
déclaré être consultant et a reçu des honoraires
d'Abbott, Orion Pharma, Protein Design Biopharma, AstraZeneca, Amgen, Takeda,
et Menarini. Le Dr Kleber a déclaré avoir reçu des
bourses de recherche de Orion Pharma et être consultant chez Abbott et
Orion Pharma. Le Dr Pocock être consultant chez Abbott, Orion Pharma, et
Scios. Le Dr Packer être consultant chez Abbott et Orion Pharma. Les Drs
Thakkar et Padley sont salariés chez Abbott. Les Drs Pöder et
Kivikko sont salariés chez Orion Pharma.
Financement/Soutien: Abbott et Orion Pharma ont financé
l'essai SURVIVE et les travaux d'analyse des données.
Rôle du sponsor: Les analyses des résultats de
l'étude ont été réalisées avec le
supervision du sponsor par ICON Clinical Research (Dublin, Ireland, et North
Wales, Pa). Le sponsor a été impliqué dans la gestion,
l'analyse et l'interprétation des données. Abbott et Orion
Pharma ont revu le manuscrit avant la soumission.
Revue statistique indépendante: Raphaël Porcher, PhD
(Department of Biostatistics, Université Paris Diderot et Hôpital
Universitaire Saint-Louis, Paris, France), ont eu un accès complet au
protocole de l'étude, aux amendements, au plan d'analyse statistique,
et aux données brutes. Selon lui, le plan d'analyse statistique est
approprié pour cette étude de même que la
présentation des résultats dans le manuscrit. A partir des
données brutes, le Dr Porcher a recalculé par informatique le
délai avant le décès au cours des 180 jours suivant le
début des traitements (critère principal) des 1327 patients
randomisés dans l'étude et a vérifié ceux
utilisés par Abbott (Abbott Park, Ill) pour l'analyse. Le Dr Porcher a
alors réalisé l'analyse principale sur la base de l'intention de
traiter de même que les analyses de sous-groupes pour la présence
d'antécédents d'insuffisance cardiaque chronique, qui
étaient prédéfinies dans le plan d'analyse statistique.
Les résultats de ces analyses sont en accord complet avec ceux
rapportés dans cet article. Le Dr Porcher a réalisé
d'autres analyses de sensibilité prévues dans le plan d'analyse
statistique ou potentiellement pertinentes, qui ont confirmé les
résultats. Une compensation financière a été
payée au Dr Porcher à l'Université Paris Diderot par
Abbott.
Comité scientifique: Alexandre Mebazaa (chair), Department of
Anesthesiology and Critical Care Medicine, Université Paris Diderot and
Hospital Lariboisière, Paris, France; Markku S. Nieminen, Division of
Cardiology, Helsinki University Central Hospital, Helsinki, Finland; Milton
Packer, Department of Clinical Sciences, University of Texas Southwestern
Medical School, Dallas; Alain Cohen-Solal, Department of
Cardiology,Université Paris Diderot and Hospital Lariboisière,
Paris, France; Franz X. Kleber, Department of Internal Medicine,
Charité Medical School, Berlin, Germany; Stuart J. Pocock, Medical
Statistics Unit, London School of Hygiene and Tropical Medicine, London,
England.
Comité de suivi des données et de la tolerance: John
Cleland (chair), Castle Hill Hospital, Cottingham, England; Dan
Langrois,CHUBrabois, Vandoeuvre-les-Nancy, France; Viatcheslav Mareev,
Cardiology Research Center, Moscow, Russia; Richard Kay, PAREXEL
International, South Yorkshire, England.
Investigateurs de l'essai SURVIVE: Autriche: Alexander Geppert
(Wilhelminenspital der Stadt Wien, Wien); Thomas Martys
(Kaiserin-Elisabeth-Spital der Stadt Wien, Wien); Johannes Mlczoch
(Krankenhaus der Stadt Wien Lainz, Wien); Jorg Slany (Krankenanstalt
Rudolfstiftung, Wien). Finland: Juhani Airaksinen (Turku University Central
Hospital, Turku); Veli-Pekka Harjola (Helsinki University Central Hospital);
Heikki Huikuri (University of Oulu, Oulu); Pirjo Mantyla (Central Hospital of
North Karelia, Joensuu); John Melin (Central Hospital of Central Finland,
Jyvä skylä); Keijo Peuhkurinen (Kuopio University Hospital, Kuopio).
France: Philippe Asseman (CHU Lille, Lille); Jean-François Aupetit (CH
Saint-Joseph et Saint-Luc, Lyon); Michel Barboteu (Centre Médical
d'Evecquemont, Meulan); Marc Benacerraf (Centre Cardiologique du Nord, Saint
Denis); Jean-Marc Boulenc (Clinique Saint Joseph, Colmar); Alain Cariou
(Hôpital Cochin Port Royal, Paris); Alain Cohen-Solal (Hôpital
Beaujon, Clichy); Pierre Coste (Hôpital Cardiologique Haut Leveque,
Pessac); Jean Luc Dubois-Rande (Hôpital Henri Mondor, Créteil);
Olivier Dubourg (Hôpital Ambroise Paré, Boulogne Billancourt);
Marc Feissel (Centre Hospitalier Belfort/Montbeliard, Belfort);
François Funck (Centre Hospitalier René Dubos, Pontoise); Michel
Galinier (Hôpital Rangueil, Toulouse); Pierre Gibelin (Hôpital
Pasteur, Nice); Yannick Gottwalles (Clinique Saint Joseph, Colmar); Louis
Guize (Hôpital Européen Georges Pompidou, Paris); Gilbert Habib
(CHU de la Timone, Marseille); Ivan Laurent (Institut Hospitalier Jacques
Cartier, Massy); Hervé Le Marec (Hôpital Nord Laennec,
Nantes-Saint-Herblain); Bruno Levy (Hôpital Central Nancy, Nancy);
Alexandre Mebazaa (Hôpital Lariboisière, Paris); Gilles
Montalescot (Hôpital Pitié-Salpêtrière, Paris);
Gérald Roul (Hôpital de Hautepierre, Strasbourg); Rémi
Sabatier (CHU Côte de Nacre, Caen); Pierre Squara (Clinique Ambroise
Paré, Neuilly sur Seine); Gabriel Steg (Hôpital Bichat-Claude
Bernard, Paris); Jean-Louis Teboul (CHU Bicêtre, Le Kremlin-Bicetre);
Paul Touboul (Hôpital Cardiologique Louis Pradel, Bron); Philippe Vignon
(Hôpital Dupuytren, Limoges); Simon Weber (Hôpital Cochin Port
Royal, Paris); Faies Zannad (CHU de Nancy, Vandoeuvre les Nancy); Robin
Zelinsky (Clinique Saint-Sauveur, Mulhouse). Allemagne: Gerhard Bauriedel
(Universitatsklinikum Bonn, Bonn); Michael Bohm (Universitatskliniken des
Saarlandes, Homburg/Saar); Michael Buerke (Klinikum der Medizinischen Fakultat
der Martin-Luther-Universitat Halle-Wittenberg, Halle [Saale]); Angelika
Costard-Jackle (AK St Georg, Hamburg); Aly El-Banayosy (Herz-u Diabeteszentrum
Nordrhein-Westfalen, Universitatsklinik der Ruhr-Universitat Bochum, Bad
Oeynhausen); Gerd Hasenfub (Universitatsklinikum Gottingen, Gottingen); Franz
Xaver Kleber (Unfallkrankenhaus Berlin, Berlin); Veselin Mitrovic (Kerckhoff
Klinik GmbH, Bad Nauheim); Thomas Munzel (Universitatsklinikum
Hamburg-Eppendorf, Hamburg); Klaus Pethig (Klinik fur Innere Medizin III der
Friedrich Schiller Universitat Jena, Jena); Andrew Remppis (Medizinische
Universitatsklinik und Poliklinik, Heidelberg); Peter Schuster (St Marien
Krankenhaus Siegen, Siegen); Robert Schwinger (Universitat zu Koln, Koln
[Lindenthal]); Ruth Strasser (Medizinische Klinik II/Kardiologie, Dresden).
Israel: Jonathan Balkin (Shaare Zedek Medical Center, Jerusalem); Tuvia Ben
Gal (Rabin Medical Center, Beilinson Campus, Petah Tikva); Daniel David (Meir
Hospital, Sapir Medical Center, Kfar Saba); Dov Freimark (Sheba Medical
Center, Tel Hashomer); Andre Keren (Bikur Holim Hospital, Jerusalem); Tiberiu
Rosenfeld (Haemek Medical Center, Afula); Yoseph Rozenman (Wolfson Medical
Center, Holon). Lettonie: Galina Dormidontova (Daugavpils Central Regional
Hospital, Daugavpils); Maija Keisa (Valmiera Hospital, Valmiera); Janis Lacis
(P. Stradina Clinical University Hospital, Riga); Alfreds Libins (Liepaja
Hospital, Liepaja); Dace Meldere (Riga Clinical Hospital Gailezers, Riga);
Jurijs Verbovenko (Riga First Hospital, Riga). Pologne: Jerzy Adamus (Central
Military Hospital, Warszawa); Marek Dabrowski (Szpital Bielanski, Warszawa);
Robert Gil (Department of Invasive Cardiology, Warszawa); Jerzy Korewicki
(Institute of Cardiology, Warszawa); Maria Krzeminska-Pakula (Bieganski
Hospital, Lodz); Grzegorz Opolski (Medical University ul Banacha 1a,
Warszawa); Wieslawa Piwowarska (Jagiellonian University School of Medicine,
Cracow); Lech Polonski (Silesian Centre of Heart Diseases, Zabrze). Russie:
Igor N. Bokarev (Moscow Medical Academy, Moscow); Nikolai A. Gratsiansky
(Hospital No. 29, Moscow); Victor A. Lyusov (Russian Medical University,
Moscow); Valentin S. Moiseyev (Russian People's Friendship University,
Moscow); Mikhail Y. Ruda (Russian Cardiology Research Centre, Moscow); Raisa
I. Stryuk, Sergey N. Tereschenko, and Vladimir S. Zadionchenko (Moscow State
Medico-Stomatological University, Moscow); Dmitry A. Zateyshchikov (Central
Clinical Hospital of Russian Government Medical Centre, Moscow). Royaume-Uni:
Sanjay Arya (Royal Albert Edward Infirmary, Wigan); Craig Barr (Russells Hall
Hospital, West Midlands); John Berridge (Leeds General Infirmary, Leeds);
Henry Dargie (Clinical Research Institute, Glasgow); Gregory Lip (University
Department of Medicine, Birmingham); Bernard Prendergast (Wythenshawe
Hospital, Manchester); Andrew Rhodes (St George's Hospital, London); Roxy
Senior (Northwick Park Hospital, Harrow); Mervyn Singer (Middlesex Hospital,
London).
Remerciements: Nous remercions Thea Nieminen, RN, Toni Sarapohja,
MSc (Orion Pharma, Espoo, Finland), et Nancy Chou, MS, RD, Leticia
Delgado-Herrera RPh, MS, Stefan Hergenroeder, PhD, Bidan Huang, PhD, Brigitte
Kalsch, MD, Udo Legler, MD, Michael Marshall, PharmD, and Greg Schulz (Abbott,
Abbott Park, Ill) pour leur rôle dans la coordination des travaux d'ICON
Clinical Research, les 2 sponsors, et le comité scientifique. Toutes
les personnes remerciées n'ont reçu aucune compensation
financière pour leur implication dans ce travail.
Affiliations des auteurs: Departments of Anesthesiologie, Critical
Care Medicine, et Cardiologie, Université Paris Diderot et
Hôpital Lariboisière AP-HP, Paris, France; Division of
Cardiology, Helsinki University Central Hospital, Helsinki, Finland;
Department of Clinical Sciences, University of Texas Southwestern Medical
School, Dallas; Department of Internal Medicine, Charité Medical
School, Berlin, Germany; Medical Statistics Unit, London School of Hygiene and
Tropical Medicine, London, England; Cardiovascular Clinical Research, Abbott
Laboratories, Abbott Park, Ill; Cardiology Unit, Clinical Research and
Development, Orion Pharma, Espoo, Finland.
BIBLIOGRAPHIE
| |
1. Cuffe MS, Califf RM, Adams KF Jr, et al. Short-term intravenous
milrinone for acute exacerbation of chronic heart failure: a randomized
controlled trial. JAMA.2002; 287:1541
-1547.
FREE FULL TEXT
2. Bayram M, De Luca L, Massie MB, Gheorghiade M. Reassessment of
dobutamine, dopamine, and milrinone in the management of acute heart failure
syndromes. Am J Cardiol.2005; 96(6A):47G
-58G.
PUBMED
3. Haikala H, Kaivola J, Nissinen E, Wall P, Levijoki J, Linden IB.
Cardiac troponin C as a target protein for a novel calcium sensitizing drug,
levosimendan. J Mol Cell Cardiol.1995; 27:1859
-1866.
PUBMED
4. Pollesello P, Ovaska M, Kaivola J, et al. Binding of a new
Ca2+ sensitizer, levosimendan, to recombinant human cardiac
troponin C: a molecular modelling, fluorescence probe, and proton nuclear
magnetic resonance study. J Biol Chem.1994; 269:28584
-28590.
FREE FULL TEXT
5. Yokoshiki H, Katsube Y, Sunagawa M, Sperelakis N. Levosimendan, a
novel Ca2+ sensitizer, activates the glibenclamide-sensitive K
channel in rat arterial myocytes. Eur J Pharmacol.1997; 333:249
-259.
PUBMED
6. Kersten JR, Montgomery MW, Pagel PS, Warltier DC. Levosimendan, a
new positive inotropic drug, decreases myocardial infarct size via activation
of K(ATP) channels. Anesth Analg.2000; 90:5
-11.
FREE FULL TEXT
7. Nieminen MS, Akkila J, Hasenfuss G, et al. Hemodynamic and
neurohumoral effects of continuous infusion of levosimendan in patients with
congestive heart failure. J Am Coll Cardiol.2000; 36:1903
-1912.
FREE FULL TEXT
8. Slawsky MT, Colucci WS, Gottlieb SS, et al. Acute hemodynamic and
clinical effects of levosimendan in patients with severe heart failure.
Circulation. 2000;102
: 2222-2227.
FREE FULL TEXT
9. Moiseyev VS, Poder P, Andrejevs N, et al. Safety and efficacy of a
novel calcium sensitizer, levosimendan, in patients with left ventricular
failure due to an acute myocardial infarction: a randomized,
placebo-controlled, double-blind study (RUSSLAN). Eur Heart
J. 2002;23:1422
-1432.
FREE FULL TEXT
10. Haikala H, Kaheinen P, Levijoki J, Linden IB. The role of cAMP- and
cGMP-dependent protein kinases in the cardiac actions of the new calcium
sensitizer, levosimendan. Cardiovasc Res.1997; 34:536
-546.
FREE FULL TEXT
11. Kaheinen P, Pollesello P, Hertelendi Z, et al. Positive inotropic
effect of levosimendan is correlated to its stereoselective Ca2+-sensitizing
effect but not to stereoselective phosphodiesterase inhibition.
Basic Clin Pharmacol Toxicol.2006; 98:74
-78.
PUBMED
12. Follath F, Cleland JG, Just H, et al. Efficacy and safety of
intravenous levosimendan compared with dobutamine in severe low-output heart
failure (the LIDO study): a randomised double-blind trial.
Lancet. 2002;360:196
-202.
PUBMED
13. World Medical Association. Declaration of Helsinki: recommendations
guiding physicians in biomedical research involving human subjects. Amended
at: the 52nd World Medical Association General Assembly; October 2000;
Edinburgh, Scotland.
http://www.wma.net/e/policy/pdf/17c.pdf.
Accessibility verified March 29, 2007.14. Publication Committee for the VMAC Investigators (Vasodilatation in
the Management of Acute CHF). Intravenous nesiritide vs nitroglycerin for
treatment of decompensated congestive heart failure: a randomized controlled
trial. JAMA.2002; 287:1531
-1540.
FREE FULL TEXT
15. O'Connor CM, Stough WG, Gallup DS, Hasselblad V, Gheorghiade M.
Demographics, clinical characteristics, and outcomes of patients hospitalized
for decompensated heart failure: observations from the IMPACT-HF registry.
J Card Fail.2005; 11:200
-205.
PUBMED
16. Zannad F, Mebazaa A, Juilliere Y, et al. Clinical profile,
contemporary management and one-year mortality in patients with severe acute
heart failure syndromes: the EFICA study. Eur J Heart
Fail. 2006;8:697
-705.
FREE FULL TEXT
17. Thackray S, Easthaugh J, Freemantle N, Cleland JG. The
effectiveness and relative effectiveness of intravenous inotropic drugs acting
through the adrenergic pathway in patients with heart failure: a
metaregression analysis. Eur J Heart Fail.2002; 4:515
-529.
FREE FULL TEXT
18. Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term
risk of death after treatment with nesiritide for decompensated heart failure:
a pooled analysis of randomized controlled trials.
JAMA. 2005; 293:1900
-1905.
FREE FULL TEXT
19. Kivikko M, Antila S, Eha J, Lehtonen L, Pentikainen PJ.
Pharmacokinetics of levosimendan and its metabolites during and after a
24-hour continuous infusion in patients with severe heart failure.
Int J Clin Pharmacol Ther.2002; 40:465
-471.
PUBMED
20. Nieminen MS, Brutsaert D, Dickstein K, et al. EuroHeart failure
survey II (EHFS II): a survey on hospitalized acute heart failure patients:
description of population. Eur Heart J.2006; 27:2725
-2736.
FREE FULL TEXT
21. Metra M, Nodari S, D'Aloia A, et al. Beta-blocker therapy
influences the hemodynamic response to inotropic agents in patients with heart
failure: a randomized comparison of dobutamine and enoximone before and after
chronic treatment with metoprolol or carvedilol. J Am Coll
Cardiol. 2002;40:1248
-1258.
FREE FULL TEXT
22. Lowes BD, Tsvetkova T, Eichhorn EJ, Gilbert EM, Bristow MR.
Milrinone versus dobutamine in heart failure subjects treated chronically with
carvedilol. Int J Cardiol.2001; 81:141
-149.
PUBMED
23. Bódi A, Szilagyi S, Edes I, Papp Z. The cardiotonic effects
of levosimendan in guinea pig hearts are modulated by beta-adrenergic
stimulation. Gen Physiol Biophys.2003; 22:313
-327.
PUBMED
ARTICLE EN RAPPORT
JAMA. 2007;297:1853.
Texte Complet
|