Strategizing the Clone Wars: Pharmacological Control of Cellular Sensitivity to Radiation
Abstract
The combined administration of ionizing radiation and systemic chemotherapy is an accepted standard of care for numerous cancers. Improved efficacy through the combination of therapies reflects several interrelated processes, including DNA damage, inhibition of DNA synthesis, alteration of cell cycle distribution, and impaired DNA repair. Insights into cellular responses to radiation have led to the use of drugs that target specific intracellular signaling pathways to sensitize cells to radiation. Combinations of chemotherapy and radiation continue to be optimized, based on preclinical and early-phase clinical data that indicate the ideal sequencing of therapies, the best combinations of agents (including radiosensitizers), and the most reliable biological markers for predicting patient responsiveness. This review summarizes our current understanding of radiosensitization as it relates to preclinical drug development and discusses the potential benefits of judiciously incorporating both traditional and targeted chemotherapy into radiation regimens.
Introduction
Despite advances in systemic cancer therapeutics over the last fifty years, cancer treatment remains a challenging task, requiring the elimination of an enormous number of tumor cells. During their lifetime, over 75% of the 1.4 million Americans diagnosed with cancer each year will receive ionizing radiation, one of the most potent therapies available to the oncologist. For over 100 years, radiation has been used to treat adenocarcinomas, lymphomas, sarcomas, and squamous cell carcinomas, in addition to a number of rare tumors. Over the last fifty years, concurrent use of both ionizing radiation and chemotherapy has improved both local control and overall survival in head and neck (1), anal (2), lung (3), and cervix cancers (4), among others. Even in cancers with a poor prognosis, such as glioblastoma, the use of chemoradiotherapy (CRT) improves survival (5).
A principal concern of oncologists is to maximize the probability of controlling tumors while limiting damage to healthy tissues, often manifested as side effects. Advances in the physical delivery of radiation have allowed physicians to irradiate tumor tissue more precisely with high doses of radiation while sparing the surrounding normal tissue. However, the laws of physics limit the extent to which normal structures can be spared and often result in decisions that favor the delivery of a low dose of radiation to large areas rather than a high radiation dose to specific regions. Compounds that differentially improve the tumor response to radiation may increase the therapeutic window of ionizing radiation (Figure 1), resulting in better patient outcomes.

Effects of drugs on radiation exposure. Probabilities of tumor control (red) and normal tissue damage (blue) can be modeled as sigmoidal dose response curves. In the left panel, these probabilities are modeled in the absence of a radiosensitizing agent. The ideal radiosensitizer (right panel) shifts the tumor responsiveness curve leftward (relative to the absence of agent, left panel) while having little effect on normal tissue; drugs that have effects on both normal tissue and tumor may shift both curves to the left (not shown).
In the 1950s, preclinical studies began to explore the benefit of using fluorinated pyrimidines to enhance the effects of radiation (6), and improved outcomes in patients with gastric and pancreatic cancer treated with 5-fluorouracil (5-FU) and radiation were reported by the 1960s (7). A true paradigm shift occurred in the 1970s when researchers at Wayne State University reported the use of pre-operative 5-FU and mitomycin C given concurrently with radiation to patients with anal cancer (8). Prior to this report, cancer therapy had often involved radical surgical approaches associated with significant morbidity, but the results of the Wayne State University research suggested that radiation and chemotherapy treatment could be given initially, followed by surgical intervention, to result in increased organ preservation, decreased morbidity, and improved quality of life. Since that time, chemotherapeutic agents have been combined with radiation in a large number of cancers with outcomes ranging from improved survival to increased toxicity (9). In 2006, the use of “traditional” chemotherapeutics along with radiotherapy was advanced by Bonner and colleagues, who reported the first trial demonstrating the ability of an antibody-based therapeutic to improve radiotherapeutic outcomes in locally advanced head and neck cancer (10). The subject of this review is the mechanistic and preclinical data supporting the use of CRT. For a more complete review of the clinical trials supporting the use of CRT, the reader is directed to several excellent reviews (11–14).
Assessing Radiosensitization
One focus of radiation biology concerns the effectiveness of ionizing radiation and interfering with the proliferation of single tumor cells into tumor cell colonies. The gold standard assay for studying clonogenesis is the clonogenic survival assay, according to which the survivability of clonogenic tumor cells (i.e., clonogens) is plotted as a function of radiation dose on a linear-log scale (Figure 2). The resultant curves are most commonly fit to a linear-quadratic equation, although a number of alternative models have been used [for a historical overview, see (15)]. Radiosensitizers are compounds that, in combination with radiation, decrease the clonogenic survival of tumor cells (as seen by a shift from the red to green curve in Figure 2). Strictly speaking, radiosensitizers are defined as compounds that can be administered at doses that sensitize cells to radiation but do not directly evoke cytotoxicity. However, in many cases, drugs have been described as radiosensitizers even though the drug itself has significant effects in the absence of radiation. The degree of radiosensitization can be expressed in terms of the sensitizer enhancement ratio (SER), quantifiable by using median effect or isobologram analyses that allow one to isolate synergy, additivity, or antagonism between two therapeutics [described in detail in (16, 17)]. Due to the compounding effect of multiple daily radiation fractions, even a small SER can have profound effects on overall tumor control (Figure 3).
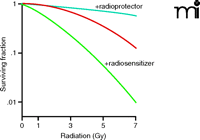
The radiation biologist’s approach to the dose response curve. Although most dose response curves are plotted on a log-linear graph, the gold standard radiobiology assay is typically represented, as shown here, on a linear-log graph, with meaningful radiation responses occurring over a range of 1 to 2 log units. Use of an agent that sensitizes a tumor to radiation results in a leftward shift (green) of the baseline radiation curve (red), whereas agents that protect cells from radiation shift the curve in the opposite direction (light blue). This analysis is modeled using a linear-quadratic formulation as Y=e−(AX+BX2).
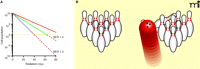
Quantifying radiosensitization and the therapeutic challenge. (A) A typical macroscopic tumor may have over 1010 cells and if even a single cell remains after treatment, the tumor may relapse. Although the benefit of a radiosensitizer may be small on any given day, the compounded effect of cell death over thirty or more days of radiation is quite large. In this example, each 2-Gy dose of radiation, administered to a tumor originally consisting of 1010 cells, kills 40% of the tumor cell population (red line). Radiosensitizers effectively decrease the dose of radiation needed to achieve a given percentage of cell death; the ratio of the required radiation dose in the absence of radiosensitizer to the (lowered) dose required in the presence of radiosensitizer is known as the sensitizer enhancement ratio (SER). Agents with SERs of 1.3 (green) and 1.7 (purple) are also modeled. Conceptually, the graph shows that a “cure” will require sufficient irradiation to decrease the number of surviving tumor cells to less than 100 (yellow horizontal line). (B) To further illustrate the difficulty of killing 10 log units of cells, imagine each bowling pin as a log of cells, with 10 bowling pins collectively representing 1010 cells. A therapy that removes 99% such a population of tumor cells (i.e., a reduction of 2 log units) is the equivalent of removing 2 pins—not a very successful frame for a bowler and certainly not a very successful therapy for a patient.
The bases on which CRT improves outcomes have been proposed to include spatial cooperation, toxicity independence, and enhanced tumor radiosensitivity (17). Spatial cooperation (illustrated in Figure 4) refers to the concept that, whereas radiation is limited to treating cancer within the target volume, chemical agents can travel throughout the body and can address subclinical metastatic disease. Combining effective systemic therapy with effective local therapy may increase the probability of cure. Toxicity independence may improve outcomes by allowing two therapies that do not have overlapping toxicity profiles to be given at near maximal doses. Finally, it is often observed that some drugs, depending on dose and time of administration, can improve the outcome of radiation. These “true” radiosensitizers are the subject of this review.

The duality of cancer and the duality of chemoradiotherapy. Most cancers require control of both local disease (large sphere) and microscopic metastatic disease (small spheres). Both radiation and chemotherapy can effectively shrink tumors. Radiation that is targeted to specific organ areas can be quite effective at eliminating disease, but it is impractical to irradiate the entire body with doses required to obliterate every cancer cell; microscopic metastases may go untreated (upper panel). In contrast, chemo-therapeutics are generally unable to eradicate primary tumors but are amenable to systemic distribution (lower left). The combination of chemotherapy and radiation is designed to improve local as well as systemic disease. The achievement of both local and systemic control is important to consider as newly developed therapies in one area (e.g., chemotherapeutics, radiation, or surgery) may affect the administration of therapies in another area.
In contrast to radiosensitizers, radioprotectors are compounds that can prevent tissue toxicity and promote clonogenic survival (Figure 2). In the ideal clinical context, radioprotection would be extended to normal cells and not to tumor cells. Although it is outside the scope of this review, the study of radioprotectors is of great interest, particularly in situations where individuals may be exposed to higher than normal levels of ionizing radiation in their work environment, such as astronauts, homeland security and military preparedness specialists, and medical professionals involved in diagnostic imaging studies [reviewed in (14)].
Platinum Compounds
Platinum compounds, such as cisplatin (Platinol), carboplatin (Paraplatin), and the third-generation agent oxaliplatin (Eloxatin), are “traditional” antineoplastic drugs (Box 1) that can be used in combination with radiation to treat a variety of solid tumors. Platinum compounds react within the cell to form DNA–protein crosslinks as well as intrastrand and interstrand crosslinks within DNA, and cellular attempts to repair these crosslinks result in strand breaks. When used in combination with radiation, platinum agents increase the number of lethal double-strand breaks in DNA (18). In addition, several alternative mechanisms have been proposed to underlie the synergistic effects of combining platinum compounds and radiation: enhanced formation of toxic intermediates via free radical formation; alteration in platinum pharmacokinetics; and inhibition of DNA repair (19–21).
Terminology: Traditional vs Targeted Drugs
The taxonomy of chemotherapeutic agents can be confusing for even experienced readers. The term “targeted” is currently used to refer to a variety of drugs, such as therapeutic antibodies, that target specific molecules. Examples of targeted drugs include tyrosine kinase inhibitors, PARP inhibitors, and mTOR inhibitors. In this way, use of the word “targeted” can be problematic, implying that “traditional” drugs, such as cisplatin and paclitaxel, which despite being well known for their specific interactions with DNA and microtubules, respectively, are somehow “untargeted.” Despite this obvious fallacy, the nomenclature of “traditional” and “targeted” will be used throughout this review.
The use of platinum drugs illustrates some of the complexities of translating work from the lab into the clinic. Bolus cisplatin, for example, has been shown to be inferior to either daily dosing or continuous infusion in mouse models of radiosensitization (22) and in at least one clinical trial (3). However, based on the higher systemic concentrations achieved with bolus dosing, along with the hypothesis that higher concentrations result in better control of metastatic disease, bolus cisplatin given every three weeks is a common clinical regimen. In addition, whereas cisplatin is often used as a single agent in head and neck and cervical cancers, carboplatin is most frequently combined with a taxane (see below) when given as a radiosensitizer, despite the fact that combinations of multiple drugs and radiation have little preclinical data supporting improved radiosensitization efficacy. Finally, oxaliplatin is thought to be a more effective radiosensitizer than cisplatin, owing to higher DNA adduct toxicity and improved inhibition of DNA synthesis in preclinical studies (23–25). Yet oxaliplatin is often combined with 5-FU for systemic therapy, and the tolerability and efficacy of this combination with radiation are still under investigation in early phase clinical trials.
Antimetabolites
Structurally similar to normal cellular metabolites, antimetabolites act as inhibitors as well as substrates of intracellular enzymes, and they cause cytotoxicity upon incorporation into nucleic acids. The folic acid analogs methotrexate and pemetrexed (Alimta) inhibit dihydrofolate reductase, an enzyme critical to DNA synthesis. Purine analogs such as 6-mercaptopurine (6-MP; Purinethol) interfere with purine synthesis and can be metabolized for incorporation into DNA, whereby they induce cell cycle arrest and apoptosis. Although some preclinical data suggest that analogs of folic acid and purines have radiosensitizing properties (26), they are less well studied than pyrimidine analogs.
Antimetabolites appear to exert their radiosensitizing effects via a combination of mechanisms. First, inhibition of deoxynucleotide triphosphate synthesis can perturb the balance of intracellular nucleosides and thereby undermines the fidelity of DNA replication. Most antimetabolites result in the arrest of cells at the G1/S boundary, a particularly radiosensitive cell cycle phase. In addition, gemcitabine and 5-FU have been reported to sensitize cells in S phase (normally a relatively radioresistant phase), although the mechanism remains unclear (27). Finally, although antimetabolites alone are unable to induce apoptosis, their effects exacerbate genomic DNA damage due to ionizing radiation and thereby initiate apoptosis.
5-Fluorouracil
Following the pioneering work by Nigro and colleagues (8), 5-FU, a uracil analog, and its oral prodrug form, capecitabine (Xeloda), remain mainstays of combined modality therapy for gastrointestinal tumors. Although 5-FU has effects on both DNA- and RNA-related processes (28), the biologic basis of its radiosensitization action is thought to be primarily via inhibition of thymidylate synthase (29), an enzyme necessary for DNA replication and repair. In addition, owing to imbalances in nucleotide pools, a metabolite of 5-FU can be deleteriously incorporated into DNA to result in single-strand breaks (30). Failure of DNA repair pathways, which is augmented by thymidylate synthase inhibition, leads to cell death. It is thus thought that radiation and 5-FU synergistically results in the inability of cells to repair the damaged DNA. When used as a radiosensitizer, 5-FU is typically given, because of its short plasma half-life, by continuous venous infusion rather than as bolus therapy (31–33). Likewise, one of the two daily doses of capecitabine is generally recommended to be taken one to two hours prior to radiation for maximal effect.
Gemcitabine
The cytidine analog gemcitabine (Gemzar) is a potent radiosensitizer, both in the lab and clinic, with activity against a variety of cancers. Gemcitabine results in the depletion of dATP pools and arrests cells at early S phase after twenty-four hours of treatment (34, 35). Radiosensitization by gemcitabine also depends on intact homologous recombination and mismatch repair processes (36, 37). Because the effects of gemcitabine (i.e., dATP depletion and cell cycle arrest) do not materialize immediately, no synergy is seen when irradiation is administered prior to the full twenty-four-hour period of drug exposure (38).
Taxanes and Microtubule Stabilization
Taxanes work by stabilizing microtubules during M phase, preventing chromosomal separation and inducing cell death [reviewed in (39)]. In vitro, cells arrested at the G2/M transition are at their most radiosensitive phase (40, 41). Synergy between paclitaxel (Taxol) and radiation appears to depend on proper sequencing and dosing of drug with respect to radiation: drug exposure prior to radiation results in supra-additive interactions (42); drug exposure after radiation results in antagonism (43); and dosing with low concentrations of drug results in merely additive effects (42). Intriguingly, the preclinical hypothesis that radiosensitization by taxanes is dependent upon G2/M arrest was not supported in a phase I trial utilizing serial tumor biopsies to investigate changes in cell cycle distribution following a dose of paclitaxel (44). The low number of patients undergoing biopsies (only five of thirty patients in the study), however, makes it difficult to assess whether the negative result accurately reflected a low radiosensitization efficacy or was rather related to experimental conditions surrounding the timing and nature of the biopsies.
Mechanisms of taxane-mediated radiosensitization may be related to a variety of factors. Milas and colleagues provided evidence in vivo that radiosensitization was dependent on the level of molecular oxygen within the tumor tissue (45). In their classic experiment, paclitaxel enhanced tumor responsiveness to radiation in mice under ambient conditions, but this enhancement was abolished when radiation was delivered after the induction of tissue hypoxia by leg clamping (45). Oxygen-dependent radiosensitization by paclitaxel appears to be related to two physiological phenomena: 1) the selective killing of aerated cells results in their removal, which then makes oxygen accessible to previously hypoxic tissue and thereby sustains the killing process; and 2) the removal of irradiated cells reduces interstitial pressure, which increases capillary flow and improves tumor oxygenation. The complex interplay of mechanisms within in vivo systems is further highlighted in that taxanes also appear to inhibit angiogenesis as well as pro-survival signaling pathways activated by VEGF and bFGF [(46); see below]. Finally, taxanes appear to promote anti-tumor immune responses that further enhance the efficacy of radiation (47).
Although paclitaxel is the taxane most commonly combined with radiation in the clinic, a number of groups have also indicated the efficacy of docetaxel (Taxotere) against head and neck cancer cells as well as prostate cancer cells (48, 49). In both metastatic settings, docetaxel has proven effective as a single agent. Docetaxel is of further interest for its alteration of radiaresponsiveness and in its toxicity against relatively radioresistant S-phase cells (50).
Topoisomerase Inhibitors
The DNA topoisomerases regulate the topology of DNA in the nucleus. The topoisomerase inhibitors topotecan (Hycamtin) and irinotecan (Camptosar), which are camptothecin analogs that inhibit topoisomerase I, as well as etoposide (VP-16, or Toposar), which inhibits topisomerase II, prevent the repair of DNA strand breaks and thereby promote cell death (51, 52). The combination of topoisomerase inhibitors and radiation can be synergistic, resulting in cell death that would otherwise not occur. In addition to inhibiting the repair of DNA strand breaks, etoposide can induce the production of reactive oxygen species that can further potentiate DNA damage (53). Finally, although the topoisomerases were initially described for their role in relaxing supercoiled DNA, topoisomerase II also helps recruit proteins, such as Rb, p53, ERK2, and Cdc2 kinase, to specific subcellular sites, which represents another dimension to the effects of etoposide in promoting cell death (54).
Reiterating a common theme, the proper sequencing of drug and radiation is necessary for optimal results with topoisomerase inhibitors. Treatment with camptothecin derivatives for two or four hours prior to radiation results in radiosensitization, whereas no effect is seen if drug treatment follows radiation (51). Increased radiosensitivity has been reported when inhibitors of both topoisomerase I and II are administered simultaneously (52), although other reports suggest that sequential treatment with topoisomerase I inhibitors followed by topoisomerase II inhibitors may be preferable to simultaneous treatment (55, 56).
DNA Alkylation in Radiosensitization
Temozolomide (Temodar), a DNA-methylating agent, results in the formation of O6-methylguanine (57) and activation of the mismatch repair pathway (58). The non-catalytic enzyme O6-methylguanine-DNA methyltransferase (MGMT) can repair the lesion and prevent apoptosis that would otherwise occur (59). Despite the initial conflicting data regarding its in vitro efficacy (60), temozolomide has been established as a radiosensitizer in glioma cell lines both in vitro and in a xenograft model, an effect that correlates with increased mitotic catastrophe rather than apoptosis or cell cycle checkpoint activation (61). Most importantly, a phase III clinical trial has warranted concurrent treatment with temozolomide and radiation as the standard of care for high-grade gliomas, a result that does not appear to depend on MGMT activity (5).
Selective Radiosensitization of Hypoxic Cells
No discussion of classic radiosensitizers would be complete without at least mentioning hypoxia-selective agents. Tumors often harbor regions of hypoxia that result from poorly organized or spatially distant blood vessels. Because molecular oxygen plays a central role in the formation of reactive oxygen species and subsequent DNA damage, hypoxic regions within tumors may often be less sensitive to radiation. Radiosensitizers designed to target hypoxic tissue have been the subject of significant in vitro, in vivo, and clinical work for nearly forty years [reviewed in (62, 63)]. Approaches to target hypoxia include the enhancement of oxygen levels in tumors, the use of oxidants that more readily diffuse into targeted tissue than can molecular oxygen, and the use of cytotoxins that are selectively active under hypoxic conditions. Unfortunately, most attempts to target the hypoxic cells within tumors have been disappointing, despite volumes of promising data from preclinical studies. Data that modestly support the use of hypoxic-cell sensitizers exist in two cases, namely, in head and neck cancer and in cervical cancer (64), but any detailed consideration of the relevant clinical trials in these and other disease states is beyond the scope of this review.
Targeting Cellular Signaling Pathways for Radiosensitization
Following a dose of ionizing radiation, reactive oxygen species are generated that affect a number of intracellular processes. DNA damage is one consequence, which activates p53, ataxia telangiectasia mutated (ATM), and ATM- and Rad3-related (ATR) proteins (65). Within minutes, activation of transmembrane receptors, including receptor tyrosine kinases (RTKs) such as the epidermal growth factor receptor (EGFR), can be detected (66, 67). Receptor activation engages downstream signaling pathways involving Ras, mitogen activated protein kinase (MEK), extracellular signal–regulated kinase (ERK), and phosphatidylinositol 3-kinase (PI3K) (68). Both the Ras/MEK/ERK pathway and the PI3K/Akt/mTOR pathway control cell proliferation, survival, and cycle progression, functions that are frequently dysregulated in cancer.
A significant body of work has shown that inhibitors of PI3K/Akt/mTOR nexus result in radiosensitization [reviewed in (69)], although the particular molecular targets within the pathway that may account for radiosensitization are not yet identified. The identification of particular signaling molecules in radiosensitization is complicated by the significant degree of crosstalk between pathways. Even so, the importance of the PI3K/Akt/mTOR pathway has been established in the radiosensitization of pancreatic cancer (70). Similarly, the Raf/MEK/ERK pathway has been implicated in the radiosensitization of breast cancer cells (71, 72) and melanoma (Janiel Shields, personal communication).
Radiation-mediated Activation of Receptors
The mechanisms by which radiation activates transmembrane receptors and intracellular signaling pathways continue to be elaborated in a wide variety of studies. It is likely that there are several cooperative processes, arising from responses to DNA damage as well as from distal consequences of ionizing events. The latter include the production of reactive oxygen and nitrogen species that can inhibit tyrosine phosphatase activities, thereby prolonging the phosphorylative activation of receptor and nonreceptor tyrosine kinases and concomitant effects on downstream signaling [reviewed in (68)]. Radiation can also activate acidic sphingomyelinase, resulting in increased production of ceramide and alterations in the clustering of receptors via changes in membrane lipid composition (73).
Over the last twenty years, several drugs that target signal transduction pathways have been approved for clinical use. A large number of additional compounds are in various stages of development. Many of these agents are investigated for their ability to target the activity of oncogenic proteins or downregulate the expression of proteins that support tumor maintenance. Investigations into a number of targeted intracellular proteins have revealed complex regulatory pathways involved in cell growth, many of which play specific roles in cellular responses to radiation and DNA damage. The following discussion will address some of these investigations as the context for understanding certain instances of “targeted” radiosensitizers (Box 1).
Epidermal Growth Factor Receptors in Radiosensitization
Overexpression of the EGFR known as ERBB1 is often associated with tumorigenesis in squamous cell carcinomas of the head and neck. This EGFR is rapidly activated following radiation (Figure 5), both in vitro and in vivo, leading to the hypothesis that its inhibition could sensitize cells to radiation. Accordingly, the development of cetuximab (Erbitux), an engineered monoclonal antibody that binds to EGFR, confirmed that blockade of the receptor in vitro could enhance radiosensitivity and promote apoptosis (74). Cetuximab, which was subsequently found to inhibit DNA damage repair and tumor angiogenesis in a xenograft model system (75), was successfully developed into a therapeutic that improves patient survival (10). This success was paralleled by the development of small-molecule inhibitors of tyrosine kinase activity, such as erlotinib (Tarceva) and gefitinib (Iressa), which have likewise shown promise as potential radiosensitizers in pre-clinical and early-stage clinical trials [reviewed in (76)].

The activation of EGFR following radiation. Transoral biopsies of soft palate cancer taken prior to (left) and four hours after (right) a dose of radiation were immunostained (brown) for the phosphorylated form of EGFR. [Protocol approved by University of North Carolina IRB (Study #08-1017).]
Similarly, the monoclonal antibody–based therapeutic trastuzumab (Herceptin), which targets human epidermal growth factor receptor ERBB2 (i.e., HER2/NEU), can overcome the radioresistance that is observed in breast cancer cell lines that overexpress ERBB2 (77). The combination of trastuzumab and radiation has shown encouraging results in a small phase II study of women with locally advanced breast cancer (78). Following up on the results of antibody-mediated blockade of EGFR receptors, the small-molecule tyrosine kinase inhibitor lapatinib (Tykerb), which has high specificity for ERBB1 and ERBB2, has been investigated in vitro as a radiosensitizer of multiple tumor types (70–72, 79). In addition, we have recently completed a phase I trial of lapatinib in women with locally recurrent, chemotherapy-refractory locally advanced, or metastatic breast cancer. Final results of the study (80), including assessments of serial tumor biopsies, are currently pending. The ideal of many such trials is to define biomarkers of responsiveness so that patient subgroups likely to derive therapeutic benefit can be identified.
Understanding aberrations in the intracellular signaling pathways (Figure 6) may allow clinicians to better predict responsiveness in patients. For example, preclinical work has recently shown that the presence of activating mutations in KRAS renders pancreatic cancer cell lines resistant to EGFR targeted radiosensitizers (70). The high prevalence of KRAS mutations in human cancers suggests that EGFR-family targeted therapies may be ineffective in a large number of patients. An important unanswered question is whether mutations in KRAS alter the efficacy of agents targeting other receptor tyrosine kinases such as platelet derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), insulin-like growth factor receptor (IGFR), and others.
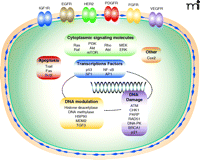
Molecular targets of radiosensitizers. Signaling molecules and pathways that have been implicated as targets in radiosensitization are schematized. See text for details.
Because Ras activation is associated with radioresistance, it follows that therapies that inhibit Ras may be effective radiosensitizers [reviewed in (81)]. Farnesyltransferase inhibitors (FTIs) block the prenylation of Ras and prevent its proper subcellular localization. Brunner and colleagues successfully used an FTI to radiosensitize activated Ras–containing cell lines in both in vitro and xenograft systems (82), although another group reported worse tumor control with an FTI and concurrent radiation in a genetically engineered mouse model driven by H-ras activation.
Angiogenesis Pathways
Tumor growth beyond a certain critical size requires angiogenesis, so that the nutrient and oxygen demands of the tumor are maintained. Growth factors that promote angiogenesis, released by endothelial cells, include basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF), all of which function by activating receptor tyrosine kinases; both antibody-based and small-molecule inhibitor approaches have been used to block angiogenesis in tumors (83). Therapy that blocks blood vessel formation, ostensibly promoting hypoxia, might well be predicted (see above) to reduce sensitization to radiation. Nevertheless, inhibitors of angiogenesis have not been shown to undermine the efficacy of CRT, prompting the hypothesis that antiangiogenic therapy may be countered by endogenous proangiogenic signals, thereby normalizing the balance of tumor vasculature responsiveness to CRT (84).
Preclinical data suggest that VEGF, bFGF, and PDGF are all upregulated in response to radiation (85), possibly reflecting activation of PI3K/Akt/mTOR and hypoxia/VEGF-induced pathways that promote cell survival and radiation resistance (see below). Indeed, endothelial cells in vitro are radiosensitized in response to the anti-VEGF-receptor–based therapeutic bevacizumab (Avastin), whereas tumor cell lines are not [reviewed in (86)], and some inhibitors of angiogenesis are synergistic with radiation treatment, both in vitro and in vivo (87). This efficacy may be a result of the general promiscuity of the kinase domain with regard to many tyrosine kinase inhibitors, a factor that is relevant to anti-VEGFR therapies. The low levels of VEGF receptor that typify some tumor cell lines suggest the possibility of direct effects on tumor cells rather than on angiogenesis. Interestingly, the downregulation of DNA repair enzymes in tumor biopsies after bevacizumab (Avastin) treatment implies that some component of the therapeutic mechanisms may be vascular-independent (88).
Modulation of Apoptosis
Two general strategies have been used to modulate the apoptotic response following radiation: 1) activation of proapoptotic cell surface receptors (i.e., the extrinsic pathway) and 2) inhibition of antiapoptotic pathways. Most of the preclinical studies utilizing apoptosis induction and radiation have been performed in hematopoetic cell lines that manifest strong apoptotic responses to radiation alone. Whether these strategies will be successful against carcinomas that lack a strong apoptotic response to radiation remains to be seen.
The extrinsic pathway of apoptosis is mediated by extracellular ligands that activate cell surface death receptors [i.e., members of the tumor necrosis factor (TNF) receptor superfamily] such as the TNF-related apoptosis inducing ligand (TRAIL) receptors, Fas/CD95, and TNF receptor 1. Because these receptors do not rely on p53-mediated apoptosis, they represent attractive targets for cancer therapy. TRAIL receptors and Fas/CD95 can be upregulated in response to DNA-damaging agents (89), providing a potential explanation for therapeutic synergy between radiation and agonists of these receptors, which include a soluble recombinant form of human TRAIL (89–91), an antibody-based agonist of TRAIL receptor 1 (92), and a recombinant Fas ligand (93). Highlighting the importance of thorough preclinical studies and consistent with the hypothesis that DNA damage upregulates TRAIL receptor expression, some groups have shown synergy in vitro only when TRAIL is added after radiation (91), whereas others have failed to replicate synergy at tolerable dose levels in vivo (93).
Antiapoptotic members of the Bcl-2 protein family, which function in part by binding and sequestering the so-called BH3 domain of proapoptotic members of the family, are frequently overexpressed in human tumors and have been associated with radioresistance. Drugs such as the phytochemical gossypol, acting as BH3 domain mimetics and antagonizing antiapoptotic interactions of Bcl-2 family members, promote apoptosis and reduce clonogenic survival in irradiated tissues (94). Similar to what has been described with respect to death receptor ligands, gossypol delivery must occur subsequent to radiation in order to manifest synergy (94, 95). Other approaches to undermine inhibitors of apoptosis (e.g., antiapoptotic Bcl-2 family members), including the use of siRNAs, antisense oligonucleotides, and ribozymes, show promise as radiosensitizers (96).
DNA Regulatory Processes as Targets in Radiosensitization
PARP Inhibitors
No group of potential radiosensitizers holds greater interest in the field of radiation biology than those that target poly(ADP-ribose) polymerase (PARP) activity. PARP-1 is induced by radiation and plays an important role in repairing single-strand DNA breaks. For nearly twenty years, drugs that affect PARP activity have been known to promote cell death in irradiated tissues by inhibiting DNA repair (97, 98). Radiosensitization appears to increase when the radiation dose is fractionated, likely reflecting the cumulative inhibition of PARP activity in early stages of DNA repair (99), and is most significant in rapidly proliferating cells, in which impairment of single-strand repair during S phase culminates in multiple double-strand breaks (100). These findings strongly suggest that PARP inhibitors may be particularly well suited for use in fractionated radiation schedules, a hypothesis currently being tested in clinical trials.
Inhibitors of Histone Modification
Histones play important roles in maintaining and regulating chromatin structure, an important component of gene regulation. Histone deacetylase (HDAC) inhibitors such as vorinostat (Zolinza) prolong histone acetylation and subsequently perturb normal regulatory processes. A variety of HDAC inhibitors, with diverse structural features, radiosensitize a variety of tumor types [reviewed in (101)]. Owing to the dynamic processes of acetylation and deacetylation, radiosensitization requires exposure to the HDAC inhibitor both before and after radiation (102). The exact mechanism of radiosensitization remains poorly understood but may be related to effects on DNA repair kinetics (103), gene transcription (104), and acetylation of nonhistone proteins (105). The phosphorylation of histones may also represent an avenue for targeted radiosensitization. Aurora B, a serine/threonine kinase essential for mitotic progression, is a chromosome passenger protein that phosphorylates histone H3 and is involved in chromosome segregation and cytokinesis. In both in vitro and in vivo experiments, the Aurora B inhibitor AZD1152 functions as a radiosensitizer via induction of mitotic catastrophe (106).
Cell Cycle Checkpoints
Following radiation-induced DNA damage, cells are transiently arrested at cell cycle checkpoints as they attempt to repair DNA lesions prior to S phase or mitosis. The cell cycle checkpoint kinases CHK1 and CHK2 integrate signals from proteins that monitor DNA damage and repair. CHK1 and CHK2 also activate effector molecules, such as p53, DNA-PK, Cdc25, PCNA, histone H3, Aurora B, and Plk1, to cause cell cycle arrest [reviewed in (107)]. Given their integrating role, checkpoint proteins make logical targets for radiosensitization, a hypothesis supported through studies of CHK inhibitors (108, 109). Specifically, it appears that blockade of CHK1 enhances apoptosis even in the presence of p53 mutations (110), whereas CHK2 has a predominant role in double-strand break repair (111). Other proteins involved upstream (e.g., ATM and ATR) or downstream (e.g., Aurora B, Rad51, DNA-PK) in checkpoint control are also potential targets for inhibitor development and radiosensitization. The importance of carefully investigating the safety of checkpoint inhibition in patients cannot be overstated, given the exquisite sensitivity of ataxia telangectasia patients to ionizing radiation (112).
Future Directions
The clinical importance of therapeutic CRT regimens and their great promise for improving patient outcomes call for significant further investigation into the mechanistic basis of radiosensitization. Despite its use in cancer therapy for over a century, the biological mechanisms underlying the effects of ionizing radiation have yet to be fully understood or pharmacologically exploited. Tremendous strides have been made in improving the technical delivery of CRT over the last thirty years, including the more frequent use of hypofractionated radiotherapy (i.e., high dose per fraction over a limited number of fractions). The optimal delivery of radiation sensitizers within this context, however, remains uncertain.
In addition, optical imaging has enabled the monitoring of individual cells during radiation, and this information is bringing advances in high-throughput techniques into radiation biology (113). These advances are slowly translating into in vivo systems, allowing for the real-time examination of responses to a variety of chemotherapy/radiation combinations as well as consideration of specific microenvironmental targets (e.g., in tumor vasculature). Because microenvironmental alterations can have significant impacts on therapeutic efficacies, it is imperative that we continue to integrate biomarkers and traditional pharmacological endpoints into studies to refine treatment approaches and improve patient outcomes.
The research and development of radiosensitizers will face particular challenges in terms of regulatory processes of drug development. The reliance on survival endpoints for regulatory approval of new compounds may be too great a burden for approval of a novel radiosensitizer, as survival depends upon a multitude of factors, of which local tumor control is only one limited issue [reviewed in (114)]. Furthermore, many drug companies view radiosensitizers as niche products, without a mass market. To date, cetuximab is the only agent designed for a particular target (i.e., the EGFR) that has been shown in phase III trials to improve survival when combined with radiation (10). However, innumerable phase I and II trials are ongoing in cancer centers across the country, and various combinations of FDA-approved small-molecule chemotherapeutics as well as newer biotherapeutics are entering into CRT. It is likely, as systemic therapy becomes better able to control micrometastatic disease, that the value of improved local control will be increasingly realized.
Acknowledgments
RJK is currently supported by a Kaye Fellowship in Head and Neck Cancer Research. He has been designated a B. Leonard Holman Pathway Fellow by the American Board of Radiology. A Resident/Fellows in Radiation Oncology Seed Grant from the American Society for Radiation Oncology supported study # 08-1017 (cited in this article.) We thank Nana Feinberg and Mervi Eeva in the UNC Anatomic Pathology Translational Core Laboratory (APTCL) for expert technical assistance. The UNC APTCL is supported, in part, by grants from the UNC University Cancer Research Fund.
- Copyright © 2010
References

Randall J. Kimple, MD, PhD is currently a Kaye Postdoctoral Fellow in Head and Neck Cancer Research and Visiting Assistant Professor in the Department of Human Oncology at the University of Wisconsin. He completed his Radiation Oncology residency at the University of North Carolina in 2010 as a Leonard B. Holman Pathway Fellow of the American Board of Radiology. He received his M.D. and Ph.D. in Pharmacology from the University of North Carolina under the guidance of David Siderovski. E-mail rkimple{at}humonc.wisc.edu; fax 608-262-7224.

