CaMKII, an Enzyme on the Move: Regulation of Temporospatial Localization
Abstract
Calcium-calmodulin–dependent protein kinase II (CaMKII) is an important regulator of neuronal and behavioral plasticity. Studies in which the subcellular distribution of CaMKII has been altered argue that targeting of this enzyme to specific subcellular compartments is crucial to many of its roles. Understanding how a very abundant enzyme can achieve specificity of action over time and space requires an understanding of the functional diversity of the enzyme and its distribution. In this review we will discuss how structurally distinct isozymes, splice isoforms, and autophosphorylation states of CaMKII can affect kinase activity and localization. We will focus on the fast activity-dependent synaptic localization of the kinase and its association with postsynaptic proteins. The ability of enzyme activation to regulate protein–protein interactions with these binding partners and the potential for such binding interactions to regulate CaMKII activity in novel ways may represent new paradigm for CaMKII regulation.
Introduction
Special challenges arise when an abundant enzyme with a broad range of substrates is required to catalyze reactions of high specificity. Calcium-calmodulin–dependent protein kinase II (CaMKII) is such an enzyme; it is present in enormous quantities in the central nervous system (1) and it has been implicated in a wide variety of neurobiological process (2–4). The key role of this kinase in synaptic plasticity and behavior has made understanding its ability to achieve precise modulation of neuronal function a timely and important problem.
CaMKII is encoded by four genes in mammals: α, β, γ, and δ. The α and β isozymes are the predominant forms found in the central nervous system (5). D. melanogaster and C. elegans each have a single gene that is alternatively spliced to produce multiple isoforms (6–8). Each gene encodes a protein that has an N-terminal serine–threonine kinase domain, followed by a regulatory region with an autoinhibitory sequence and a calmodulin- (CaM)-binding site. The C-terminus is usually called the association domain and is responsible for assembly of subunits into large (estimates range from 8 to 14 subunit) multimers (2, 9).
Aside from its great abundance in the brain, the feature of CaMKII that caught the imagination of neuroscientists was its ability to act as an autophosphorylation-regulated molecular switch. Binding of Ca2+-CaM to the regulatory domain of the kinase activates the kinase by releasing the catalytic domain from inhibition by autoregulatory sequences proximal to the CaM binding site. This allows the kinase to both phosphorylate itself and its substrates. Autophosphorylation of T286 in α CaMKII and in C. elegans CaMKII (T287 in mammalian β, δ, and γ CaMKII and in D. melanogaster CaMKII) occurs fast and renders the kinase Ca2+/CaM-independent, although the activity of this form of the kinase is less than the Ca2+-CaM bound form. Interestingly, T286 “autophosphorylation” occurs in trans within the holoenzyme; both the “substrate” and “kinase” subunits must have CaM bound. Phosphorylation of T286 also changes the affinity of the kinase for CaM, decreasing the off rate by a factor of a thousand (10).
Once the kinase is Ca2+-independent, secondary sites (T305 and T306 in α CaMKII and T306 and T307 in β CaMKII, D. melanogaster and C. elegans) that are within the CaM binding site can be phosphorylated. These sites are inaccessible in the presence of CaM, but when they are phosphorylated they block CaM binding. Phosphorylation of T305 and T306 acts to prevent further stimulation by calcium, resulting in a catalytically inactive kinase if T286 is unphosphorylated or a partially active kinase in the presence of phosphoT286. As will be discussed, the autophosphorylation and isozyme-specific structural properties of CaMKII provide important mechanisms for regulating its localization.
From the time of its discovery, it was apparent that CaMKII was present in many cellular compartments. Subcellular fractionation studies revealed that CaMKII associates with both the soluble cytosolic fraction and cell membranes (11). Interestingly, the α/β heteromultimer enzyme from these two fractions appears to be identical, in terms of their physical properties, substrate specificity, pH dependence, Kact for CaM, and Km for synapsin I (12). Subsequent studies suggested that there is an α CaMKII-directed association with synaptic structures and that CaMKII is a major constituent of the postsynaptic density (PSD), a proteinaceous structure present on the cytoplasmic side of the membrane at the site of synaptic contact (13). A high α to β ratio (3:1) correlates with greater PSD association of CaMKII in rat forebrain compared to cerebellum where the ratio is 1:4 and there is less CaMKII in the PSD (5, 14). In cultured hippocampal neurons, the amount of CaMKII associated with the PSD increases as the α to β ratio increases during synaptogenesis (15). It is important to note, however, that the amount of CaMKII associated with the particulate fraction also correlates with how the kinase is isolated. For example, ischemia during tissue preparation was shown to lead to α CaMKII translocation to the PSD (16, 17).
These biochemical studies suggested that CaMKII is a synaptic protein and, more importantly, that its association with the synapse is regulated and dynamic. In this review we will examine the strategies used by neurons to target CaMKII to tissues, subcellular compartments, and protein complexes.
Localization of CaMKII to Specific Tissues and Subcellular Compartments
Biochemical studies demonstrated that CaMKII is found in many cell types and subcellular compartments. In this section, we discuss the non-synaptic distribution of CaMKII.
Tissue-Specific Expression of CaMKII Isozymes and Alternatively Spliced Isoforms
One mechanism by which the activity of CaMKII can be adapted to the needs of an individual cell is by tissue-specific expression of a particular CaMKII isoform. As noted above, mammals have four genes encoding highly related members of the CaMKII family: α, β, γ and δ. Differences in structure between isozymes occur primarily within discrete variable regions (18). All four genes undergo alternative splicing in these variable regions and the resultant enzymes can have distinct biochemical properties. For example, β binds to actin (19) and has a 10-fold higher affinity for CaM compared to that of α (20). The α CaMKII gene has an alternatively spliced form, termed alpha CaMKII association protein (α KAP), which contains only the association domain and a domain that facilitates binding to the sarcoplasmic reticulum (21). This form can make heteromultimers with full-length kinase and target the kinase to the sarcoplasmic reticulum. The α, δ, and γ isozymes all have alternative splice forms that contain nuclear localization signals (21 –23). In Drosophila, where there is only a single CaMKII gene, alternative splicing also gives rise to enzymes with different affinities for CaM and substrates (24–26).
CaMKII isozymes are not uniformly expressed in time and space; α and β are found primarily in nervous tissue with β expression initiating during embryonic development (22) and α postnatally (27). The α isozyme is the primary CaMKII in the forebrain, whereas β is the primary cerebellar isozyme (5). β is also found in glia (28). The γ and δ isozymes are found at low levels in all tissues (29) with enrichment of various splice forms in particular non-neuronal tissues.
Alternative splicing of CaMKII genes is also regulated in time and space. Splicing of the β isozyme generates embryonic forms that are not seen in the mature nervous system (22). Splicing of the α gene to produce a form that contains a nuclear localization signal is observed in thalamus and hypothalamus, but not in caudate putamen (22). These expression and splicing mechanisms allows catalytically and structurally distinct isoforms of the kinase to be targeted to specific cell-types within a tissue at particular developmental times.
Nuclear Localization
The molecular weight of the CaMKII holoenzyme, estimated to be between 400–600 kDa, likely prohibits passive diffusion of the enzyme into the nucleus; nonetheless, in early studies, nuclear CaMKII activity was observed (30). More recent molecular cloning studies have found that some α, γ, and δ alternatively spliced isoform sequences contain a nuclear localization signal (NLS) in their variable regions. In the αB and δB isoforms, the NLS has been shown to be functional (21–23), whereas in the γA isoform, activity of the NLS may be suppressed by other sequences in the variable region (31). The presence of CaMKII in the nucleus gives it access to additional substrates that can affect cell function. In ventricular myocytes, the δB isoform regulates expression of atrial natriuretic factor (32), and the expression of the α isoform in the nucleus of PC12 cells prevents NGF-induced neurite extension (33).
Nuclear entry of these CaMKII isoforms is regulated by several mechanisms (Figure 1⇓). The multimeric nature of CaMKII and its ability to form heteromeric multimers (5) suggest that isoforms containing an NLS would coassemble with cytosolic subunits that could possibly retard its translocation. Consistent with this idea, Srinivasan et al. found that the ability of CaMKII to enter the nucleus correlated with the ratio of nuclear/cytoplasmic subunits within a holoenzyme (21). Thus, in any given tissue, the ability of CaMKII to enter the nucleus is a function of the levels of all the expressed CaMKII isoforms, not just the one(s) containing an NLS.
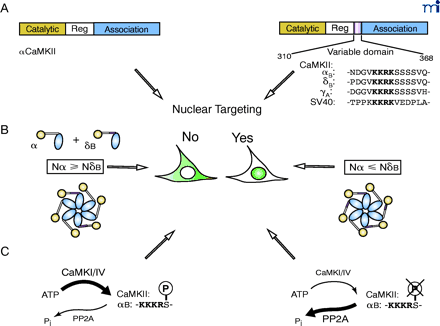
Mechanism for nuclear targeting of CaMKII. A. CaMKII isoforms that contain a nuclear localization sequence (NLS) target nuclear entry. The prototype NLS of SV40 T antigen is included for sequence comparison (21–23). B. The ratio of cytosolic isoform vs. nuclear isoform determines the subcellular localization of δB CaMKII (21). C. Phosphorylation close to the NLS by CaMKI or CaMKIV prevents nuclear entry of αBCaMKII (23), whereas an increased level of PP2A was associated with nuclear targeting of δBCaMKII (91).
Nuclear entry of CaMKII can also be regulated by other kinases. In all mammalian nuclear CaMKII isoforms, the NLS is followed by a string of four serine residues. Phosphorylation of the first serine by CaMKI or CaMKIV blocks both nuclear translocation of αB and binding of CaMKII to pendulin, an importin-α subunit (23). Phosphorylation of T286 in the regulatory domain of CaMKII may also affect nuclear localization (34) ; a nuclear form of a T286-specific phosphatase has been identified (35).
Additional non-NLS mechanisms may also exist to allow CaMKII into the nucleus. In Drosophila, CaMKII immunoreactivity is present in nuclear extracts and nuclear staining is observed immunohistochemically (Long, Hodge, and Griffith, unpublished results), but the genomic sequence does not predict any isoforms that might contain a canonical NLS. CaMKII in this organism is also SUMO-conjugated, a posttranslational modification often associated with nuclear proteins (36). In summary, nuclear targeting of CaMKII is controlled by factors including cell identity and the activity of other signal transduction pathways.
Cytoskeletal Localization
Cytoskeletal proteins were the first candidates in the search of CaMKII anchoring proteins (37–39). Indeed, hippocampal pyramidal neurons treated with actin- and microtubule-depolymerizing agents exhibit a reduction in PSD-associated CaMKII (40). As to the specific cytoskeletal network components involved, F-actin, α-actinins, densin-180, and myosin V all bind to CaMKII in vitro and in complex with other proteins in presynaptic vesicles or the PSD (Table 1⇓). Of this group, myosin V is interesting in that it has a role in movement of vesicles and organelles, and mice harboring myosin V mutations have nervous system defects (41). Other proteins associated with the PSD will be discussed in more detail in the section on localization of synaptic CaMKII. The association between CaMKII and the actin-based motor protein myosin V (42) makes it plausible for CaMKII to shuttle between cytosol and PSD in dendritic spines along the actin filament network. In the case of cell cycle control, it was demonstrated that myosin V-mediated organelle transport is aborted by CaMKII phosphorylation of myosin V in response to mitotic stimuli (43). Whether CaMKII has a role in regulation of myosin V function in the nervous system remains to be determined.
Proteins that Interact with CaMKII
Targeting of CaMKII to the actin cytoskeleton is a function of the β isozyme. β CaMKII binds directly to F-actin and copurifies with cellular actin (19). Ca2+-CaM disrupts the interaction of β with actin (44, 45), allowing neuronal calcium levels to regulate association. Coassembly of α CaMKII with β allows it to be targeted to the actin cytoskeleton. Only a small fraction (approximately 15%) of holoenzyme subunits must be β (19). Studies using chimeric α/β subunits have established that the actin binding activity of β CaMKII maps largely to its variable domain (45). Alternative splicing in the variable domain of β CaMKII can also affect the level of actin association, because β’ was found to be less effective in actin targeting than β (19).
One function of the actin targeting ability of β CaMKII is regulation of neurite motility and branching. In relatively young, < 5 d.i.v. (days in vitro), cultured hippocampal neurons, β< but not α, promotes motility and arborization of dendrites (45). In more mature cultures, > 13 d.i.v., β no longer causes branching, but rather increases synapse formation. This suggests that over time the context in which the cytoskeletally targeted CaMKII acts changes. Studies with point mutants and CaMKII inhibitors indicated that, although CaMKII activity is required for these effects, the enzymatic activity of the β subunit itself is dispensable. It is possible that coassembly of β CaMKII with α CaMKII contributes to its morphological effects (45). In general, the ability of transfected CaMKII subunits to modulate motility correlated more highly with their ability to bind actin than with their enzymatic activity.
Localization to the Sarcoplasmic Reticulum in Muscle
In skeletal muscle, β and δ isoforms of CaMKII are tightly associated with the sarcoplasmic reticulum (SR) (46). In this tissue, the α CaMKII gene is transcribed from an internal intronic promoter instead of the neuronal 5′ promoter elements. The transcripts generated lack coding sequences for the catalytic domain and are spliced to contain an exon encoding a novel N-terminal hydrophobic sequence proximal to the association domain (47, 48). Two splice forms of this transcript have been characterized and differ in that one has an NLS and the other a CaMKII phosphorylation site (49). These splice forms, called α KAP, are integral membrane proteins that associate with the sarcoplasmic reticulum membrane fraction of the skeletal muscle. α KAP colocalizes with a Src Homology 3 (SH3) domain-containing splice form of β CaMKII, βM, and several δ splice forms in the SR membrane (46). By binding to α KAP, CaMKII remains tethered in close proximity to its important skeletal muscle substrates, including the integral membrane protein phospholamban, the ryanodine receptor, and the sarcoplasmic reticulum Ca2+-ATPase (SERCA) pump.
Localization of Synaptic CaMKII
The documented role of CaMKII in learning and memory suggest that theF synaptic localization of the kinase allows it to carry out important functions. In this section, we will consider the mechanisms by which CaMKII levels and isozyme ratio at the synapse can be modulated and the time scales over which these processes occur.
Localization of Camkii Synthesis
Activity-dependent regulation of synaptic CaMKII levels
One mechanism that allows CaMKII to be responsive to cellular needs is through the regulation of enzyme levels by synaptic activity. Local changes in synaptic activity that lead to local changes in CaMKII concentration or isozyme type could allow the kinase to take on new functions at an active synapse. This type of regulation occurs over a long (hours for mRNA synthesis and transfer) to medium (minutes for local synthesis) time scales.
The amounts of and ratio of α CaMKII and β CaMKII are regulated by activity in a number of neuronal cell types and tissues. Stimulation of hippocampal slices acutely increases both autophosphorylation at T286 and the level of total CaMKII immunoreactivity (suggesting increased amounts of CaMKII) (50). The increase in α CaMKII immunoreactivity is due to de novo synthesis in dendrites (51). Changes in synaptic activity in multineuronal pathways in vivo can also modulate kinase levels. In macaque visual cortex, monocular deprivation leads to increases in α CaMKII mRNA while levels of β CaMKII mRNA decrease in the deprived ocular dominance column; γ and δ mRNA levels remain unchanged (52). Total CaMKII immunoreactivity increases with deprivation (53). In these studies the nature of the activity change in the cortical neurons studied was not assessed. Activity also appears to increase α CaMKII levels in rat visual cortex. Dark-reared rat pups that are exposed to light rapidly polyadenylate α CaMKII mRNA and synthesize α CaMKII protein (54). These findings are consistent with a study by Bagni et al. in which they demonstrated that chemical stimulation of synaptosomes with glutamate–glycine or KCl increased the association of polyribosomes with α CaMKII mRNA (55). In hippocampal cultures, bidirectional modulation of α and β has been demonstrated. For example, application of TTX to suppress neuronal activity results in a fast (1 h) decrease in α CaMKII levels and a slower (3 h) increase in β CaMKII levels (56). Treatment of the cultures with bicuculline, which increases overall activity by suppressing inhibitory transmission, causes changes in the opposite direction: increased amounts of α and decreased β. The modulation of the α/β ratio in visual cortex and hippocampus may serve as a homeostatic mechanism to adjust the sensitivity of the holoenzyme to calcium by altering the proportion of β CaMKII, which is more sensitive to Ca2+-CaM.
The increase in CaMKII mRNA level and, presumably, translatability, suggests that much of the increase in CaMKII protein is due to new synthesis. It is possible, however, that steady-state synaptic kinase levels might also be influenced by the local rate of degradation. In a study of the effects of activity on PSD composition, Ehlers found that although there was an increase in the amount of α CaMKII in PSDs of bicuculline-treated hippocampal cultures and a decrease at synapses in TTX-treated cultures, the half-life of the protein did not change (57). This was in contrast to other PSD components such as the N-methyl-D-aspartate receptor (NMDAR) subunits NR1 and NR2A, where abundance changes were accompanied by changes in protein half-life. These data suggest that the major regulator of CaMKII levels is new synthesis.
Mechanisms of dendritic targeting and translation of αCaMKII mRNA
Targeting of α CaMKII mRNA to dendrites and de novo protein synthesis clearly contribute to local activity-dependent increases in synaptic α CaMKII levels (Figure 2⇓). Synaptic activity is capable of regulating local translation, as noted above, but it can also regulate transport of the α CaMKII mRNA to dendrites (58). Transport of the α CaMKII mRNA is controlled by elements within the 3′ UTR of the message (59). β CaMKII mRNA apparently does not contain such signals, because it is not found in dendrites (27). Two elements have been identified in the α message: 1) a 30-bp proximal element that targets the mRNA to the dendrite, and 2) a distal element that represses transport unless the neuron is active (60). The identity of the repressing element is unknown, but that region of the message contains a cytosolic polyadenylation element (CPE), and such elements have been implicated in mRNA transport (61).
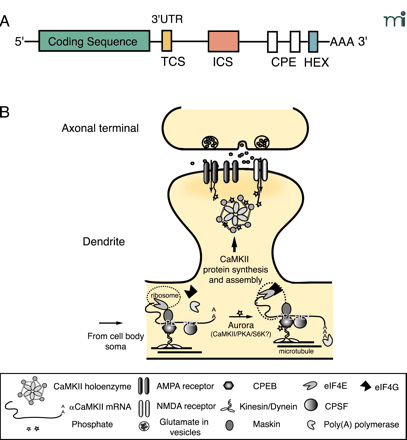
Mechanisms for dendritic targeting and translation of α CaMKII mRNA. A. The mRNA sequence structure of αCaMKII. The 3′ UTR of αCaMKII has several elements that are important for dendritic targeting. The Targeting Cis-Sequence (TCS) is a 27-nt sequence that contains a RNA transport sequence (CGCAGAGAUC) known to be necessary for the targeting of myelin basic protein mRNA in oligodendrocyte. The 648-nt Inhibitory Cis-Sequence (ICS) requires synaptic activity to override its intrinsic inhibition of dendritic targeting. There are two Cytoplasmic Polyadenylation Elements (CPEs)—UUUUUAU—in the 3′ UTR of CaMKII. The hexanucleotide (i.e., AAUAAA) polyadenylation signal (HEX) is shown. These elements can bind to trans-acting proteins, such as the CPE-binding protein CPEB, which has been implicated in facilitation of the targeting of αCaMKII mRNA (54, 59, 60). B. Regulatory mechanisms for the dendritic targeting and translation of αCaMKII mRNA. Transient synaptic activity leads to transport of αCaMKII mRNA from the soma to the base of the dendrite where transcripts associate with ribosomes through an interaction between CPE and CPEB. Motor proteins (i.e., Kinesin/Dynein) shuttle the protein–transcript complex on the microtubule network to the dendrites (61). CPEB also binds to maskin, which has an inhibitory interaction with the eukaryotic initiation factor eIF4E at the cap of αCaMKII mRNA and prevents translation until triggered by synaptic activity. Sustained synaptic activity activates Aurora—a Ser–Thr kinase whose activity might be required for local translation of dendritic mRNAs—or possibly other kinases such as PKA, CaMKII or S6 kinase to phosphorylate the different isoforms of CPEB in synapses (63, 92). Phosphorylation of CPEB triggers a conformational change that brings the HEX-binding cleavage and polyadenylation specificity factor CPSF in contact with CPEB. Maskin is released from eIF4E which then recruits eIF4G for proper assembly of αCaMKII mRNA onto ribosomes. Poly(A) polymerase extends the poly-A tail and nascent CaMKII polypeptides are synthesized (92).
Local translation of α CaMKII mRNA is controlled by CPEB, a trans-acting factor that binds to the CPE in the 3′ UTR (54). Binding of CPEB to the CPE holds mRNAs in a form that is inefficiently translated. Activation of CPEB by phosphorylation drives polyadenylation of α CaMKII mRNA and allows it to be translated by dendritic ribosomes (62). The kinase(s) responsible for CPEB activation in the nervous system are unknown, but several CPEB genes are expressed in the brain with consensus sites for a number of different signal transduction pathways (63). Activity-dependent polyadenylation of α CaMKII mRNA has been demonstrated in visual cortex (54), and synaptic synthesis of a green fluorescent protein (GFP) reporter flanked by CaMKII UTRs was demonstrated in dendrites that had been mechanically isolated from their soma and treated with brain-derived neurotrophic factor (BDNF) (64).
There are several possible functional consequences of local translation of α CaMKII mRNA. The most obvious is that at synapses where synthesis occurs, the local concentration of enzyme will increase. The nature of this additional enzyme, however, is unusual: it is an α homomultimer. This form of the kinase has the unique ability to bind to densin-180, a transmembrane protein that is associated with the PSD (65). β CaMKII and α/β heteromultimers do not associate with this protein. This may allow the newly synthesized kinase to participate in macromolecular complexes from which CaMKII synthesized on somatic ribosomes is excluded. This newly synthesized α homomultimer would also be expected to fail to bind to dendritic actin because only β CaMKII contains actin-binding sequences (19). The alteration of the α/β ratio by local translation may push the neuron into a less morphologically, but more functionally—in terms of synaptic strength—plastic regime. This is consistent with findings that α CaMKII overexpression stabilizes dendritic trees (66), whereas overexpression of β CaMKII stimulates their arborization (45). Overexpression of constitutively active α CaMKII in cortical pyramidal cells leads to selective retention of only active synapses and a net loss of inactive connections, thus activated α CaMKII may also have an additional role in synapse elimination (67). These studies suggest that the activity and the isozyme profile of neuronal CaMKII are key factors in sculpting neuronal networks.
Dynamic Localization of Synaptic CaMKII
Visible synaptic accumulation of GFP-tagged α CaMKII occurs within seconds (44). As compelling as the evidence for local protein synthesis to be an underlying mechanism of synaptic accumulation is, the time scale over which it occurs is too long to completely account for CaMKII appearance in PSDs. The particle containing CPEB, CPE-containing mRNA, and motor proteins moves at 4–8 μ/min on microtubules (61). The average distance to travel from the cell soma to the far end of dendrites in a hippocampal pyramidal neuron is over 30 μ. At this speed, it would not be feasible to transport CaMKII mRNA to the dendrites, initiate local translation, and produce visible CaMKII accumulation in dendritic processes within the first seconds after of onset of synaptic activity. Even just the translation step, which in eukaryotes occurs at a rate of about seven amino acids per second, would take over a minute for α CaMKII. This implies that additional mechanisms for fast synaptic localization of presynthesized CaMKII exist. In this section, we will discuss visualization of this fast localization and some of the biochemical mechanisms that might underlie translocation.
Visualization of CaMKII translocation
Studies done in cultured hippocampal CA1–CA3 pyramidal neurons by Meyer’s group provided the first real-time visualization of fast CaMKII translocation to synaptic sites (44). Genes encoding GFP-tagged CaMKII subunits were expressed and the subcellular distribution of the GFP signal monitored in real time. Translocation of GFP-tagged CaMKII was induced by tetanic stimulation or application of glycine–glutamate. GFP–CaMKII went from a diffuse distribution in the cytoplasm of the dendrite to a punctate localization at regions where there appeared to be cell–cell contacts. Colocalization with PSD95 and FM 4-64 staining was consistent with the puncta being synaptic sites. Accumulation of CaMKII at these sites was reversible and depended on external calcium and NMDAR activation. The use of the GFP-tagged kinase allowed this group to obtain detailed kinetic information about CaMKII synaptic association and dissociation.
These elegant real-time imaging results are consistent with other studies in which the movement of CaMKII into the synapse was visualized post facto by histochemical methods. Brief depolarization or exposure to glutamate–glycine (1.5 min) causes rapid thickening of the PSD, as visualized by electron microscopy, in hippocampal slices (68). The thickening was rapidly reversible with replacement of the solution with the control saline. Immunoelectron microscopy revealed that there was an increase in CaMKII levels coincident with the increased thickness of PSD.
Activation of PKC can also regulate association of CaMKII with synaptic sites. Craig’s group found that treatment of rat hippocampal cultures with phorbol esters caused a movement of CaMKII into synaptic sites (69). The kinetics of this translocation was slower than that observed for GFP–CaMKII after glutamate–glycine treatment, with half maximal translocation seen at ten minutes. Translocation was accompanied, on the same timescale, by a movement of NMDAR out of the synaptic region. In this instance, movement of the endogenous kinase was measured by immunohistochemical methods and translocation was qualitatively different compared to that observed with GFP-CaMKII. Phorbol ester–dependent redistribution of CaMKII did not require the activation of NMDAR, voltage-gated calcium channels, or synaptic activity. One manipulation that did block kinase translocation was dissolution of the neuronal F-actin network with latrunculin A. PKC-dependent CaMKII translocation might, therefore, occur by a completely different mechanism than the NMDAR-dependent movement of the kinase studied by other groups. The slow time course of PKC-dependent CaMKII accumulation at synapses could imply that both new synthesis and translocation are involved in the generation of CaMKII puncta in these experiments. The loss of immunoreactivity in the dendritic cytoplasm, however, argues that at least part of the accumulation is due to translocation.
Fast redistribution of CaMKII in an intact organism has also been demonstrated. In zebrafish, stimulation of the skin activates primary sensory afferents that have glutamatergic synapses onto a commissural interneuron, CoPA. Both stimulation of the skin and direct application of glutamate caused cytosolic GFP–CaMKII to become punctate in the CoPA interneuron (70). As in hippocampal cultures, translocation was fast (maximal in less than a minute), reversible and required activation of NMDAR. The use of a natural stimulus in an intact organism is convincing evidence that endogenous calcium signals can trigger CaMKII redistribution.
The role of autophosphorylation in synaptic targeting
Point mutants in GFP–α CaMKII were used to probe the requirements for autophosphorylation and CaM binding in fast NMDAR-dependent translocation (44) (Figure 3⇓). CaM binding was required for translocation, as a point mutant in the CaM-binding domain (A302R) was unable to move to the synapse in response to neuronal stimulation. Enzymatic activity, tested by mutation of the ATP-binding site of the kinase (K42R), and T286 phosphorylation, tested by phosphomimetic and “dephosphomimetic” mutants T286D and T287A, respectively, were not required for association, but T286 phosphorylation affected the off-rate of the kinase (71). Dissociation of T287A was more rapid than wild type. In contrast, the dissociation half-time of the T286D mutant increased from twelve seconds to over three minutes. One speculation as to the mechanism for this failure to dissociate is that CaMKII autophosphorylated at T286 has a much higher affinity for CaM (10), and thus remains in the CaM-bound state much longer.
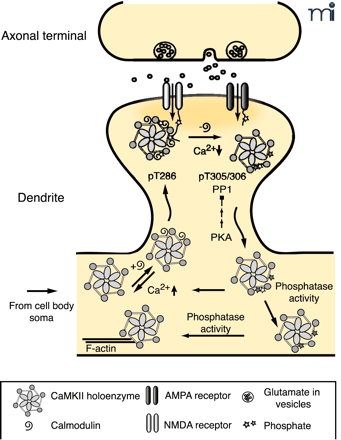
The role of autophosphorylation in dynamic activity-dependent translocation of CaMKII to the PSD. Synaptic activity leads to NMDA receptor activation and Ca2+ influx allowing Ca2+-CaM to bind to CaMKII promoting translocation to the PSD. Autophosphorylation at T286 (bright star) traps CaMKII in the PSD until Ca2+ levels decrease and CaM dissociates from CaMKII. CaMKII autophosphorylated at T305, 306 (dark star) is unable to rebind CaM, leading to dissociation of CaMKII from the PSD. PP1 activity, which can be overcome by increased PKA activity, dephosphorylates pT286 and facilitates the dissociation process. The cytosolic pool of CaMKII exists as a mixed population of kinases, with respect to their different states of autophosphorylation. Those with residual pT286 are primed to translocate to the PSD rapidly with a lower amplitude synaptic stimulus, whereas kinase with pT305/306 requires cytosolic phosphatase activity to return to the basal state (19, 44, 71).
β CaMKII also accumulates at synapses, but the kinetics of translocation of CaMKII containing subunits of this isozyme is different from α CaMKII. α CaMKII reached half maximal translocation in twenty seconds, whereas α/β heteromultimers and β CaMKII were much slower—eighty and 280 seconds, respectively (44). Although the F-actin binding of β CaMKII might have been expected to account for these slower kinetics, Meyer’s group demonstrated that the release from the actin cytoskeleton was very fast, on the order of five seconds, and therefore, was not the major factor slowing the transport of β-containing holoenzymes. Release of β CaMKII from F-actin was facilitated by both the presence of calcium and autophosphorylation at T287, but translocation of β was still slower than α. One possible explanation for the difference may be that β-containing holoenzyme has different, less kinetically favorable, binding sites in the PSD compared to the α homomultimer.
The ability of T286 phosphorylation of α CaMKII to modulate the length of its association with the synapse suggests that the rate of dephosphorylation of this site is likely to be an important regulatory factor (72, 73). Consistent with this, inhibition of protein phosphatase 1 (PP1), which is thought to be the major PSD phosphatase that acts on T286 (74), caused prolongation of α CaMKII association (71). Application of 8Br-cAMP, a cAMP-dependent protein kinase (PKA) activator, to the cultured neurons also decreased the off-rate, perhaps reflecting the ability of PKA to turn down PP1 activity by activation of phosphatase inhibitors.
Autophosphorylation within the CaM-binding domain of CaMKII at T305 and T306 can also affect synaptic localization of the kinase. These sites are available only when CaM has dissociated from the kinase and phosphorylation there is known to block CaM binding and CaM-dependent activation. Phosphorylation of these residues in vitro decreases the ability of CaMKII to bind to isolated PSDs (73). For GFP–CaMKII in hippocampal cultures, mutation of these threonines to non-phosphorylatable alanines (T305A/T306A) drastically decreased the dissociation rate from synaptic sites (71). Recently, the Silva group generated knock-in mice that express only T305D or T305V/T306A α CaMKII (75). In the T305D mice, the amount of α CaMKII in the PSD fraction was greatly decreased, as was T286 phosphorylation. Although the overall levels of β CaMKII were unchanged, the amount of β in the PSD was also reduced. This appeared to be a dominant effect of the T305D α CaMKII, because in α CaMKII-null animals, the amount of β in the PSD actually increases. Ironically, this means that the overall level of CaMKII at the PSD is higher in α CaMKII-null animals than in T305D knock-ins. T305D animals had profound LTP defects even compared to the α CaMKII null animals, perhaps reflecting this difference in overall kinase levels. In animals carrying the T305V/T306A mutations, the amount of both α and β kinase in the PSD was increased by about 50%. Measurements of LTP and behavioral plasticity in these animals suggested that these sites are important for setting plasticity thresholds; T305V/T306A mice had a lower threshold for LTP, but impaired spatial learning.
Alterations in phosphorylation of T286 and T305 that affect CaMKII localization are also seen as a downstream consequence of a seemingly unrelated mutation. In the mouse model of Angelman Syndrome (AS), there is a maternal deficiency of E6-AP ubiquitin ligase, an enzyme involved in protein degradation. Humans and mice with this deficiency have a characteristic spectrum of problems including ataxia, seizures, and learning disability. Investigation of the plasticity defects in AS mice revealed abnormal LTP that appeared to be arise from faulty signal transduction downstream of Ca2+ influx (76). An exhaustive analysis of the known kinase pathways that influence generation of LTP found that levels and phosphorylation of PKC isoforms, PKA subunits and ERKs were all normal. Levels of CaMKII were also normal, but T286 and T305 autophosphorylation was significantly increased. Catalytic activity of CaMKII was decreased, presumably due to a decreased sensitivity to Ca2+-CaM of enzyme phosphorylated at T305. The amount of CaMKII associated with the PSD in AS mice was about 50% of normal, consistent with the T305D knock-in study that indicated that phosphorylation of T305 dominantly decreases PSD binding (75). The proximal cause of the altered phosphorylation of T286 and T305 in AS mice appears to be a change in phosphatase activity. Both PP1 and PP2A are capable of dephosphorylating T305 in vitro (77). In AS mice, the protein levels of both phosphatases are normal, but activity is significantly decreased (76). The cause of this change in phosphatase activity is unknown.
The knock-in and AS mouse data on T305 phosphorylation and PSD association have at least two possible mechanistic interpretations. One possibility is that CaM is necessary for binding and stable association of the kinase with the PSD. In this instance, the mutation or phosphorylation of T305, 306 would act to modulate CaM’s ability to bind. A second possibility is that the CaM-binding region of the kinase has other partners in the PSD and that these interactions may also be regulated by phosphorylation. An example of such a protein is Drosophila Camguk, the fly homolog of mammalian CASK. Camguk associates with Drosophila CaMKII and, in the absence of Ca2+-CaM and T287 autophosphorylation, promotes the phosphorylation of T306, 307 on the kinase. Phosphorylation results in the dissociation of the membrane-bound complex, and inactive autophosphorylated kinase is released (C.S. Lu et al., in press).
Because both T286 and T305, 306 autophosphorylation play roles in the activity-dependent translocation of CaMKII to the PSD, multiple distinct states of the kinase’s competency for translocation might exist depending on the ratio of the autophosphorylation of T286 to T305, 306. In fact, a “primed” state of translocation was proposed—in addition to “basal” and “synapse-bound”—based on the observation that GFP–α CaMKII that has just dissociated from PSD can be induced to translocate with a lower amplitude stimulus (71). It was speculated that this primed translocation was due to partial dephosphorylation by PP1 activity in the PSD. Another implication of the two-site regulation of translocation is that the turnover rate among the different states of CaMKII on-the-move may be regulated by changes in local phosphatase activity.
Synaptic Binding Partners of CaMKII
The dynamic redistribution of CaMKII that occurs with synaptic activity is likely due to changes in protein–protein interactions. The number of identified binding partners for CaMKII has grown rapidly in the last few years (Table 1⇑). In this section, we will discuss a subset of the activity-dependent synaptic interactors.
One of the first PSD proteins identified as a CaMKII binding partner was densin–180 (38, 78), a membrane associated, synaptic protein (79). Its ability to bind to CaMKII and to act as CaMKII’s substrate are regulated by its alternative splicing (78). Densin-180 is capable of binding unphosphorylated α CaMKII, but phosphorylation at T286 (65, 78) or T286 and T305, 306 (78) enhances binding. Only the α isozyme is capable of binding directly to densin-180, although densin-180 can interact indirectly with β by binding to α-actinin, another PSD protein that can associate with α/β heteromultimers (65). This trimeric complex links both somatically and dendritically synthesized CaMKII to the actin cytoskeleton.
CaMKII can bind to subunits of the NMDAR (80 –82), which, clustered at postsynaptic sites opposing presynaptic neurotransmitter release sites, function as molecular coincidence detectors, requiring both glutamate binding and depolarization-mediated removal of Mg2+ block to allow channel opening. The NMDAR is critical for development, synaptic transmission, learning, and memory. NMDA receptors are heteromultimers, consisting of an essential NR1 subunit and combinations of NR2A-NR2D and NR3 subunits (83). Binding of CaMKII was initially reported to differing combinations of subunits: NR1 and NR2B, but not NR2A (82), NR2A and NR2B (80) and NR2B, but not NR2A or NR1 (81). Differences in binding conditions and in the intrinsic affinities of the interactions are likely responsible for these discrepancies.
Binding to NR1 requires autophosphorylation of CaMKII at T286 but is not affected by autophosphorylation at T305, 306 (84). Ca2+-CaM in the absence of ATP does not enhance association. The CaMKII binding domain on NR1 (aa 843–863) overlaps NR1’s CaM and α-actinin2 binding sites. Accordingly, these three proteins compete for occupancy of the NR1 C-terminal, setting up a situation that allows CaMKII binding to initiate a cascade of structural rearrangements in the PSD.
Binding of CaMKII to NR2A may be an order of magnitude weaker than binding of CaMKII to NR1 or to NR2B (81, 84). Association of CaMKII with NR2A (aa 1349–1464) requires autophosphorylation, and is not enhanced by Ca2+-CaM in the absence of ATP (85). This binding is partially competed by a truncated α CaMKII (aa 1–315) suggesting that interaction occurs with CaMKII’s catalytic or autoinhibitory domain. The C-terminal region of NR2A also contains a sequence that binds to PSD95. Binding of CaMKII and PSD95 was found to be competitive both in vitro and in coimmunoprecipitations from hippocampal slices (85). In a subsequent study, the same group found that phosphorylation of S1416 by PKC could negatively regulate CaMKII binding (86). This finding is particularly interesting because PKC-dependent loss of synaptic NMDARs coincides with increased amounts of synapse-localized CaMKII (69). Dissociation of CaMKII would be necessary to prevent it from leaving the synapse with the NMDAR. These results again point to CaMKII binding as being a catalyst for structural change that may be regulated by other signaling pathways.
The binding of CaMKII to NR2B is equally interesting and complex. Two sites in the C-terminal of NR2B have been defined: the C site (aa 1289–1310) and the P site (aa 839–1120). Association with the P site, like binding to NR1 and NR2A, requires autophosphorylation of T286, but is not affected by Ca2+-CaM alone or phosphorylation of T305, 306 (84). Interaction of CaMKII with the C site is promoted by Ca2+-CaM binding to the kinase in the absence of autophosphorylation (87, 88). This interaction occurs in a region of NR2B that contains a CaMKII phosphorylation site: S1303. The impact of phosphorylation of this site toward binding CaMKII is controversial. Colbran’s group found an inhibition of association of the T286 autophosphorylated kinase with this region when S1303 was phosphorylated and an increase in dissociation rate (87). Schulman’s group found no increase in dissociation, although it is unclear what time scale they examined (88).
Both groups agree that this region of NR2B interacts with the catalytic domain of CaMKII. Bayer et al. showed that this interaction, in addition to localizing the kinase to the plasma membrane, locked it into an active conformation that was independent of Ca2+-CaM or T286 autophosphorylation (88). This bound kinase could remain associated with the NMDAR in the presence of low calcium concentrations and was refractory to autophosphorylation of T305, 306. The mechanism of activation rests with the ability of a small region of NR2B to mimic the autoinhibitory domain of CaMKII. When the kinase is activated, either by Ca2+-CaM binding or by T286 phosphorylation, the autoinhibitory domain is displaced from the catalytic domain. This generates a new binding surface on the catalytic domain that NR2B can use. NR2B then serves to keep the catalytic site open and allows the kinase to remain active even when calcium in the cell falls.
Conclusions
Does subcellular targeting of CaMKII matter? Based on studies in which the mRNA localization (89), the nuclear localization (32), or the PSD localization were manipulated (75) the answer has to be affirmative, although the explicit role of fast translocation of CaMKII has not been addressed. Altering CaMKII’s ability to visit certain parts of the neuron can result in very substantial functional changes. These studies argue that localization is a critical component of how CaMKII regulates neuronal activity and that it must be considered when positing a role for this enzyme in a process.
What does targeting do for the cell? Isozymes and alternatively spliced isoforms can have different catalytic properties, for example, Km for substrate or Kact for CaM. Differential localization allows these differences to be exploited to generate specificity of action. Localization of the kinase to a complex containing a substrate can also have profound consequences for the rate of the phosphorylation reaction. In a complex with kinase and substrate bound in a fixed ratio, assuming the substrate is available to the kinase, phosphorylation can be triggered quickly by activation of the kinase because there is no diffusion step. For a given substrate molecule, the reaction becomes independent of the cellular concentrations of kinase and substrate. Conversely, if the kinase is in a complex in an orientation that is not favorable for it to act on a potential substrate, that substrate can be shielded from fast phosphorylation. Preformed complexes can therefore impart speed and specificity to particular reactions.
What is the identity of the PSD protein responsible for the fast binding of CaMKII to the PSD? In studies with GFP–CaMKII, translocation required Ca2+-CaM and was enhanced by, but not dependent on, T286 phosphorylation (44). These studies and others (75, 76) also pointed to T305, 306 phosphorylation as being important for dissociation from the synapse. The PSD docking site(s) for CaMKII must therefore have binding properties that mirror these requirements. CaMKII has Ca2+-dependent interactions with a variety of synaptic proteins (Table 1⇑). In some cases, the biochemical mechanism has not yet been determined, so the relative roles of CaM and autophosphorylation are unknown. A number of investigators have put the NMDAR forward as a candidate for the docking site. Biochemical studies with NR2B are nicely consistent with the GFP studies, although the interactions with NR2A and NR1 are not. It is also not clear that the number of NMDAR in a single synapse is sufficient to account for the large amount of kinase that relocates. The fact that PKC-dependent translocation of CaMKII to the synapse is accompanied by a loss of synaptic NMDAR (69) implies that, at least in this case, the docking site is not the NMDAR. The large number of potential interactors present in the PSD, and the possibility of multiple translocation mechanisms, makes identification of a single binding site unlikely.
Is there a general mechanism for activity-dependent CaMKII binding? In many cases, proteins that interact with CaMKII do so only in the presence of Ca2+-CaM or autophosphorylation (Table 1⇑). The generation of novel interaction sites on CaMKII through conformational changes associated with enzyme activation may be a common theme among CaMKII binding proteins. Binding of Ca2+-CaM and ATP, and T286 autophosphorylation can all cause changes in the interaction between CaMKII’s catalytic and autoinhibitory domains. Each of these changes may create a new surface with which the CaMKII binding proteins can interact. The NR2B subunit of the NMDAR, Eag, a Drosophila voltage-gated potassium channel, and the L-type calcium channel all associate with CaMKII by mechanisms that involve interaction with a part of the catalytic domain that is not available in the inactive kinase (88, Sun et al. submitted, 90). Camguk, as mentioned above, interacts with the autoregulatory domain of CaMKII, but the interaction is promoted by ATP binding to the catalytic domain (Lu et al., in press).
A number of these activation-specific interactors also affect the activity of CaMKII. This should not be too surprising because they are binding to the catalytic and autoregulatory surfaces that control kinase activity. NR2B and Eag activate the kinase, whereas Camguk causes autophosphorylation-dependent inactivation. Given the number of proteins that interact with the kinase in a Ca2+-dependent manner, it is likely that additional proteins that can modulate CaMKII activity through direct binding interactions are going to be identified, and these interactions may play a key role in facilitating the specificity of CaMKII action.
The multiplicity of binding mechanisms also predicts that there will be no consensus sequence that can predict if a protein will bind to CaMKII in an activity-dependent manner. In two cases—the NR2B subunit of the NMDAR (88) and the Eag potassium channel (Sun et al., submitted)—the CaMKII-binding domain of the partner shares sequence similarity with the CaMKII autoinhibitory domain and interacts with the catalytic domain causing activation. In other cases, no obvious homology exists, or the binding occurs to a different part of the kinase. Further studies on CaMKII interactors and their ability to influence CaMKII activity will undoubtedly shed light on these remaining questions and raise many interesting new ones.
Acknowledgments
This work was supported by grants to L.C.G. from the National Institutes of Health: GM54408, NS44232 and MH067284. Andy Hudmon and S. Lynn Bostrom provided helpful comments.
- © American Society for Pharmacology and Experimental Theraputics 2010
References

Xiu Xia Sun, PhD, (bottom right) is a postdoctoral fellow at Brandeis University.

Cecilia S. Lu, BS, (center right) is a graduate student in the Neuroscience PhD program at Brandeis University.

Leslie C. Griffith, MD, PhD, (top right) is an Associate Professor of Biology and member of the Volen National Center for Complex Systems at Brandeis University. Address correspondence to LCG. Email: griffith{at}brandeis.edu; fax 781-736-3107.

