Targeting PGE2 Receptor Subtypes Rather Than Cyclooxygenases: A Bridge Over Troubled Water?
Prostanoids mediate a variety of cellular interactions in physiological and pathological processes, including hemostasis and thrombosis; glomerular filtration and water balance; ovulation, embryo implantation, and development; initiation of labor or abortion; and inflammation and modulation of immune responses. Prostanoids are biologically active metabolites of arachidonic acid (AA). In response to different stimuli (i.e., physical, chemical, hormonal, cytokines, etc.), AA is mobilized from membrane phospholipids through the action of phospholipases (PL) and is then converted to prostaglandin (PG)H2 by cyclooxygenase (COX)-1 or COX-2 (Figure 1⇓). COX isozymes catalyze a two-step reaction, first cyclizing AA to form PGG2 and then reducing the 15-hydroperoxy group to form PGH2. Cell-specific isomerases or reductases catalyze the conversion of PGH2 to biologically active end-products including PGE2, PGF2α , PGD2, PGI2 and thromboxane (TX) A2, known collectively as prostanoids (Figure 1⇓). Prostanoids are produced as needed in the cell of origin, act primarily as autacoids on the parent cell and/or neighboring cells, and have very short half-lives.
COX-1 and COX-2 isozymes catalyze the same reactions, show approximately 60% identity in their amino-acid sequence within a given species, but are encoded by two different genes, located in different chromosomes. They seem to have different functions even within the same cell type (1, 2): COX-1-dependent prostanoids serve a number of physiologic “housekeeping” functions, such as modulation of platelet aggregation and cytoprotection in the gastrointestinal mucosa (1). In addition, the expression of COX-1 is developmentally regulated in many different tissues including thymus (1, 3), and small changes in expression (e.g., 2- to 4-fold increase) can occur after stimulation with hormones or growth factors (1). In contrast, COX-2 is induced in macrophages, fibroblasts, vascular endothelial cells, and smooth muscle cells by various cytokines, endotoxins, growth factors, or tumor promoters (2). Therefore, COX-2-dependent PGs play a major role in inflammation and cell proliferation; however, constitutive expression of COX-2 has been found in certain regions of the brain, reproductive tissues, kidney, and thymus (2, 3).
For many years, the rate-limiting step in the production of PGs was thought to be the activation of PLs to release membrane-bound AA (Figure 1⇓). The discovery of COX-2 in the early 1990s and of the different mechanisms through which the two COX isoforms are regulated added a new rate-limiting step in PGs biosynthesis through the inducible conversion of AA by COX-2 (Figure 1⇓). Subsequently, it became evident that the protein expression of specific down-stream PG synthases, such as the PGE2 synthases (PGES), could be induced, leading to an overall increase in enzymatic activity PGE2 production (4, 5). Two isoforms of PGES have been described––one membrane-associated (mPGES) and one cytosolic (cPGES) (4, 5)––and COX-1 and COX-2 exhibit differential “functional” coupling to each isoform of the PGES in different cell types, including platelets (4–7). There has been no demonstrated physical binding between COX and PGES; rather, it is more likely that intracellular colocalization of these enzymes creates a local zone where AA is processed by physically close (but not coupled) enzymes. In addition, intracellular distribution of both COXs and PGES appears compartmentalized, in that COX-2 and mPGES are mainly membrane-associated and localized in the perinuclear envelope (6). Intracellular compartmentalization and functional coupling create preferential and selective “routes” for AA in response to different biological demands. Indeed, PGE2 and PGI2 appear to be the main prostanoids produced by COX-2, whereas COX-1 can generate all of the PGs (6, 7), depending on the availability of other enzymes present. Finally, the complex regulation and tissue specificity of prostanoid actions is enriched by a variety of cell receptors which trigger different intracellular signaling (8).
PG receptors were first characterized pharmacologically and classified based on their sensitivity to five primary prostanoids (i.e., PGE2, PGI2, TXA2, PGD2 and PGF2α ), and termed EP (for, E type prostanoid receptor), IP, TP, DP, and FP, respectively (8). Among prostanoids, PGE2 has the most receptors: four subtypes of EPs have been characterized so far: EP1, EP2, EP3, and EP4, defined on the basis of their pharmacological profiles (8). EPs are encoded by distinct genes and have divergent amino-acid sequences, but all bind PGE2 with higher affinity than other prostanoids. Thus, based on multiple receptor subtypes, PGE2 can trigger several different intracellular signal transduction paths and has diverse final effects, which sometimes seem to be even functionally opposing within the same cell or organ.
Activation of the EP1 receptor most likely increases intracellular Ca2+, through Gq, phospholipase C (PLC)/inositol triphosphate signaling, and protein kinase C (PKC) activity. EP1-mediated Ca2+ increases, however, might not be solely dependent on Gq activity (8). EP2 and EP4 stimulate adenylate cyclase via Gs, leading to the production of adenosine 3′ ,5′-monophosphate (cyclic AMP, cAMP), which then activates the cAMP-dependent protein kinase (PKA) (9). Stimulation of EP4 is also known to activate phosphoinositide 3′-kinase (PI3K) (9). At variance with other EPs, the EP3 has multiple splice variants, each having a unique C–terminal cytoplasmic tail (8), which adds more complexity to EP3-mediated signaling. EP3 generally inhibits adenylate cyclase through the activation of Gi (a pertussis toxin-sensitive G protein); however, EP3 is likely to signal through G-protein–Rho interactions as well (8).
The complexity of the final response to PGE2 is further complicated by evidence that multiple EPs are often coexpressed or induced in the same cell or organ. The biological significance and regulation of this coexpression is currently unknown, but it surely indicates that the response to PGE2 is tightly modulated and hardly predictable, based on the activation of different pathways by different EP subtypes. The “plasticity” of the final PGE2-triggered response in a given cell, may also vary depending on the local PGE2 concentration. The diversity of receptors, of intracellular signaling, and the possible coexpression of more than one isoform altogether call for the need of extensive investigation on PGE2 to assess its final biological effects and the metabolic pathways in which it is involved in different cell types and tissues. The pleiotropic effects of PGE2 are reflected in the complexity of phenotypes generated by the disruption of each EP in mice (Table 1⇓) (10–28). Overall, data from knockout animals further confirm that more than one EP is often involved in the same physiological or pathological process (e.g., immunity, inflammation, cancerogenesis, or brain damage) with different or even opposing contributions.
Given the extraordinary complexity, fine regulation, and tissue specificity of the final effects of the ubiquitous system of prostanoid––and of PGE2 in particular––it is not surprising that blocking the total PGE2/prostanoid production in each organ, through the pharmacological inhibition of upstream COX-1 and/or -2, might potentially be beneficial and deleterious at the same time. Aspirin (ASA) and non-steroidal anti-inflammatory drugs (NSAIDs), which are among the most widely used agents worldwide, inhibit COX activity, preventing the formation of prostanoids. Individual NSAIDs show variable potencies against COX-1 compared to COX-2, although none of them shows greater than 20-fold preference for COX-2. Highly selective COX-2 inhibitors, called Coxibs, have been marketed in late nineties and showed comparable anti-inflammatory and analgesic activities with fewer gastrointestinal complications than traditional NSAIDs (2). This therapeutic profile is compatible with the inhibition of COX-2-dependent PGE2 synthesis, which is involved in inflammation and pain, as previously shown by in vitro, animal, and human studies; however, Coxibs have been associated with a 2- to 3-fold excess of cardiovascular events (29–32). Although the extent of this excess needs further definition, and ad hoc placebo-controlled studies are lacking, this phenomenon appeared influenced by the pre-existing profile of a patient’s cardiovascular risk (29–32). The hazardous cardiovascular effect mediated by the Coxibs likely arises from inhibition of COX-2-dependent PGI2 released from endothelium [which is protective for vessel injury (33)] without a concomitant inhibition of COX-1-dependent platelet activation. Alternatively, it may be an indirect effect of increased blood pressure (34), or it might be related to the inhibition of COX-2 dependent anti-inflammatory pathways (35). It is unknown whether this profile applies to Coxibs only or to NSAIDs as well, for clinical trials versus placebo are lacking. Furthermore, as shown by the Therapeutic Arthritis Research and Gastrointestinal Event (TARGET) study (36), the excess of cardiovascular complications was nonsignificant in short-term studies (37). Regardless of the final outcome of this issue, altogether, these data strongly encourage the development of new therapeutic strategies [beyond a generic COX-1/COX-2 inhibition, either selective (Coxibs) or non-selective (NSAIDs)], that possess higher specificity and selectivity toward EP subtypes in different organs and disease states. Although widely used, NSAIDs possess serious gastrointestinal, renal, hemostatic, and cardiovascular side effects as well (38), which, again, arise from the unselective blockade of COX-1 and COX-2 in different organs. Therefore, the development of not only better coxibs but also better NSAIDs is needed.
One possible route toward better therapeutics is provided by the role of COX-2-dependent PGE2 synthesis and of different EPs in models of ischemic stroke. Several data on murine models of ischemic injury largely show a proischemic effect of COX-2. COX-2 is overexpressed in ischemic lesions (in neurons, glia, and vessels) of rodents and humans, with inflammatory cells invading the lesion (39). In particular, COX-2 appears to be expressed at the synaptic site of glutaminergic neurons and contributes to neurotoxicity mediated by N-methyl-D-aspartate (NMDA) glutamate receptors (40,41). These receptors play a major role in lesions derived from stroke, seizures, and neurodegenerative diseases (42). COX-2 gene deletion or selective COX-2 inhibition in wild-type mice attenuates NMDA-mediated neurotoxicity and ischemic brain injury (43,44). This effect appears to be mediated by PGE2 (43). Recently, Doré and colleagues demonstrated a role for EP1 in promoting NMDA-related neurotoxic events (28). In fact, specific EP1 activation caused a greater area of brain damage (approximately 30% increase) in mice after NMDA treatment as compared to that observed in controls; these lesions were reduced in EP1-null mice or by an EP1 antagonist given to wild-type animals. On the other hand, the same group had previously reported that selective activation of the EP4 subtype is protective against NMDA-induced neuronal damage (20). Also, EP2 activation reduces lesions in models of cerebral ischemia (17); therefore, EP1 antagonists and EP4/EP2 agonists might constitute useful agents for the acute treatment of stroke, in spite of (and instead of) COX-2 inhibition.
Another interesting field for targeting EPs rather than COX-2, is atherosclerosis. Atherosclerosis is a chronic process in humans, where early (i.e., fatty streaks) and late lesions (i.e., stable or ulcerated plaques) have substantial histological and physiopathological differences. Both pro- and anti-atherogenic effects of COX-2 have been described in animal or human plaques (45). The anti-atherogenic role of COX-2 relies on the endothelial release of protective, COX-2-dependent PGI2. On the other hand, COX-2-dependent PGE2 release from resident macrophages in human plaques might exert a proatherogenic action, as these cells may be involved in the rupture of the plaque and thus, in the generation of a symptomatic cardiovascular event (i.e., infarction) (45). In fact, production of matrix metalloproteinase (MMP)-2 and MMP-9, enzymes capable of degrading the extracellular matrix, has been shown to occur in macrophages through a PGE2–cAMP-dependent pathway (46). Increased expression of active MMP-2 and MMP-9 has been reported in vulnerable regions of unstable human carotid plaques that contain macrophages (47,48). Thus, localized increases of COX-2 and of PGE2-dependent MMPs in resident macrophages might cause plaque disruption. Furthermore, EP4 overexpression is associated with enhanced inflammatory reactions in human atherosclerotic plaques, suggesting a contribution of the EP4 receptor to plaque destabilization (49). Recently, Pavlovic et al. have directly proved that in macrophages, COX-2-dependent MMP expression is effectively blocked by the administration of selective EP4 antagonists or by silencing EP4 expression via small interfering RNAs (siRNA) (50). Therefore, in patients with advanced atherosclerosis, targeting the EP4 might be a better strategy than COX-2 blockade in preventing symptomatic plaque rupture.
So far, only few prostanoid receptor agonists or antagonists have been tested or are currently used in human disorders. Thromboxane receptor (TP) antagonists have been studied for their utility in preventing cardiovascular disorders. The potential advantages of potent TP antagonists compared with low-dose aspirin are related to the discovery of aspirin-insensitive agonists of the platelet receptor, such as TXA2 derived from the COX-2 pathway and 8-iso-PGF2α (an F2-isoprostane that is a product of the free radical–catalyzed peroxidation of arachidonic acid) (51). The latter can synergize with subthreshold concentrations of other platelet agonists to evoke a full response, thus amplifying platelet activation in those clinical settings associated with enhanced lipid peroxidation (51). The TP antagonist, S-18886 (terutroban) is currently being compared to low-dose aspirin in a large randomized trial (PERFORM) in patients with a recent cerebrovascular event [i.e., ischemic stroke and transient ischemic attack (TIA)] (52). Iloprost and other PGI2 analogs are used in pulmonary arterial hypertension (53). Among EP agonists, misoprostol, a non-selective EP2/EP3/EP4 agonist, is used systemically as gastroprotective agent (54) and both locally or systemically in obstetrics (off-label use) for termination of pregnancy (55), induction of labor (56), and prevention of postpartum hemorrhages (57). Lower abdominal pain, diarrhea, chills, and fever are the most common side-effects (up to 90%) of sublingual misoprostol, which is associated with better bioavailability than oral administration. These multiple side-effects might reflect the activation of several EPs by this poorly selective EP agonist. An intravenously administered selective EP2 agonist, ONO-8815Ly, has been studied in healthy women as an inhibitor of uterus contractions (58), but little else has been reported on the utility of EP2 agonists in preventing uterine contractions. Thus, the clinical pharmacology of targeting prostanoid receptors in humans is still very limited. There might be several reasons for this, including: 1) prostanoids are unstable and short-lived compounds, therefore they cannot be directly used in clinical settings; 2) of all the prostanoid receptors, the EP receptors have been the last to be cloned, thus their pharmacological characterization and clinical application are still under development; 3) given the extraordinary complexity of effects of PGE2, careful experimentation should be performed on humans to assess the net response to each agonist or antagonist through systemic and local administration; and 4) the timing and length of administration should be carefully considered. Nonetheless, more refined research on the targeting of prostanoid receptors may very well increase the specificity and decrease the side-effects associated with pharmacological regulation of the prostanoid pathways in human disorders and may lead to more promising therapeutic options (as opposed to direct COX-2 inhibition) in the future.
The Pleiotropic Action of PGE2: Phenotypes of EP Knockout Mice
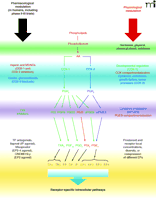
Arachidonic acid cascade and its physiological and pharmacological modulation. The metabolism of arachidonic acid to prostanoids is represented. Physiological regulations (right side of the figure) and pharmacological modulators (left side of the figure) are shown at different steps of the pathway. Enzymatic pathways, products, regulation, and drugs preferentially mediated by COX-1 are indicated in green, whereas items preferentially mediated by COX-2 appear in blue. Substrates, modulators, products, or drugs common to both isoenzymes are shown in red. See text for detailed explanations of abbreviations.
Acknowledgments
This viewpoint is dedicated to the memory of Dr. Nicola Maggiano, great scientist and friend. The critical reading and much appreciated suggestions of Prof. Carlo Patrono are gratefully acknowledged. Supported by a FIRB grant from the Italian Ministry of University (RBNE01A882_005) and by the “EICOXANOX” grant (project number LSHM-CT-2004-005033).
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Bianca Rocca, MD, PhD, is Adjunct Professor of Pharmacology at the 2nd Medical School of the University of Rome “La Sapienza,” Italy. She received her MD from the Catholic University of Rome, Italy, in 1989; her PhD from the University of Pisa, Italy, in 1995; and her Board Certification in Haematology from the Catholic University of Rome, Italy, in 1995. She was a Postdoctoral Fellow from 1995 until 1998, at the Department of Pharmacology of the University of Pennsylvania, Philadelphia, PA, directed by Prof. Garret A. FitzGerald. From 1999 until 2004 she was a Research Associate and Clinical Fellow at the Research Center of Physiopathology of Haemostasis at the Catholic University of Rome. E-mail: b.rocca{at}tiscali.it, fax : +39 0871 541 261.

