sorLA: Sorting Out APP
- Sanjiv Shah and
- Gang Yu
Although a century has passed since its initial case description, Alzheimer Disease (AD) remains a debilitating disease with little therapeutic intervention. Beginning with small failures in memory that gradually become more noticeable, AD eventually progresses to irreversible memory loss and cognitive dysfunction. In this most common of neurodegenerative disorders, the cerebral accumulation of amyloid-β (Aβ) peptides remains a major causative agent (1, 2). Recent evidence suggests that synaptic and neuronal toxicity mediated by soluble, oligomeric Aβ––as opposed to Aβ monomers and fibrils––may correlate more closely with the initial onset of cognitive impairment (3). Notable are also the findings that immunization and the resulting Aβ clearance lead to reduction of amyloid burden and slowing of memory loss (4–6). Thus, controlling Aβ generation and accumulation remains at the forefront of therapeutic approaches aimed towards AD.
Proteolytic processing of the amyloid precursor protein (APP) results in the production of various Aβ peptides (Figure 1⇓). Central to APP processing is its cleavage by either ectodomain shedding protease, α-secretase or β-secretase, followed by a remarkably unusual intramembrane proteolytic event performed by the γ-secretase complex. Ectodomain sheddases––such as the metalloproteases tumor necrosis factor–α cleaving enzyme (TACE)/A Disintegrin and Metalloprotease (ADAM)17, ADAM9, ADAM10, and metalloproteinase/disintegrin/cysteine-rich protein 9 (MDC-9), and the aspartyl protease β-site APP-cleaving enzyme 2 (BACE2)––are candidates implicated in α-secretase activity (7). α-Secretase cleaves APP approximately sixteen amino acid residues within the Aβ start site, thus mitigating the formation of Aβ. Alternatively, amyloidogenic processing of APP by the aspartyl protease BACE1 (β-secretase) creates the N terminus of Aβ (8–10). The membrane-tethered fragments of APP become immediate substrates for γ-secretase activity that cleaves heterogeneously within the transmembrane region of substrates (11).
The essential components required forγ-secretase activity include presenilins (PS1 or PS2) (12), nicastrin (13), orthologs of the C. elegans anterior pharynx defective-1 (APH-1a or APH-1b) (14), and presenilin enhancer 2 (PEN-2) (15) (Box 1). The major genetic causative factors for early-onset familial AD, the presenilins, contain two critical membrane-imbedded aspartates that provide the catalytic activity of γ-secretase (16). Although γ-secretase-mediated APP processing remains an area of dominant interest in AD research, a number of other cell-surface receptors are cleaved within their respective transmembrane domains by this enzyme, including the release of Notch intracellular domain, which is critically involved in cell fate determination. The broad substrate selectivity of the γ-secretase complex can be explained by nicastrin’s ability (within the complex) to recognize the free α-amino moiety of the (many) immediate γ-secretase substrates that are generated by ectodomain shedding of APP and other type I transmembrane proteins (17). The immediate substrates of γ-secretase are products of β-secretase and other ectodomain sheddases discussed in the previous paragraph.
The Composition of the γ-Secretase Complex
There are two presenilins (PS1 and PS2) and two APH-1 (APH-1a and APH-1b) isoforms. Each of the presenilins and each of the APH-1 isoforms are present in separate γ-secretase complexes. Therefore, four distinct γ-secretase complexes can exist in humans.
The secretases are obvious drug targets of intense interest in pharmaceutical and academic labs. The realization that a broad range of substrates are recognized and subsequently cleaved by γ-secretase, however, raises concerns of unintended toxicity resulting from the total inhibition of γ-secretase activity. The current goal of inhibiting γ-secretase is to find a therapeutic window or a way where Aβ production is inhibited without impairing essential processes associated with the other γ-secretase substrates. On the other hand, inhibiting β-secretase is expected to have less adverse effect because knocking out BACE1 revealed that this gene is not physiologically essential, although the development of potent β-secretase inhibitors presents its own challenges. In light of these considerations, alternative biochemical pathways involved more specifically in the regulation of APP processing may reveal novel targets for inhibition of Aβ generation. In this regard, the recent identification of sorting protein-related receptor/lipoprotein receptor 11 (sorLA/LR11) as a multidomain neuronal sorting factor for APP exposes a new and intriguing step in the sorting of APP from amyloidogenic versus nonamyloidogenic processing (18).
The importance of sorLA/LR11 in AD was proposed by Scherzer et al. when lymphoblasts and brain tissues from donors were shown to exhibit greatly reduced amounts of this mosaic neuronal type-I membrane protein, which contains multiple homologous domains of members of the LDLR (low-density lipoprotein receptor) and vacuolar protein sorting 10 protein (Vps10p) families (19). Extending this important observation, Anderson and colleagues have now tested the hypothesis that sorLA/LR11 may be part of a biochemical pathway that regulates APP trafficking and processing (18). In a series of intriguing experiments using in vitro, cell based, and knockout mouse models of sorLA/LR11 function, they show that this mosaic neuronal receptor interacts with APP in a manner that affects its trafficking and proteolytic processing. Surface plasmon resonance and analytical ultracentrifugation approaches revealed stoichiometric (1:1) direct binding of the ectodomains of sorLA/LR11 and APP; however, the exact regions of the ectodomains responsible for binding and whether known binding ligands can compete this interaction remain to be elucidated. Cell-based overexpression and immunoprecipitation in the presence of cross-linkers confirmed this interaction. Colocalization of the two was also observed by performing fluorescence lifetime imaging (FLIM), a relatively sensitive fluorescence resonance energy transfer (FRET)-based technique. Remarkably, sorLA/LR11 overexpression in cells and neurons resulted a shift in localization of APP to the Golgi with concomitant reduction in the amount of secreted Aβ, which is likely generated in the endocytic compartments. Expression of a cytoplasmic-domain-deleted sorLA/LR11, which accumulates on the cell surface, revealed an increase of APP cell-surface staining, indicating that sorLA/LR11 participates in the trafficking of APP to a particular subcellular compartment.
Trafficking and targeting of APP to a particular compartment may influence amyloidogenic versus nonamyloidogenic processing (20, 21). A small portion of APP molecules that do make it to the cell surface are believed to be preferentially cleaved by α-secretase. Thus, enhancing trafficking of APP to the cell surface may favor a nonamyloidogenic pattern of proteolysis. Cell surface APP that is endocytosed and delivered to late endosomes or those residing in the late Golgi and trans-Golgi Network (TGN) are predominantly cleaved by β-secretase. γ-Secretase activity has been detected in multiple compartments including a small portion that is localized at the cell surface. The amyloidogenic processing mainly occurs in the TGN and endocytic compartments as APP is trafficked through the secretory and recycling pathways. Thus, Anderson et al. propose that sorLA/LR11 overexpression may trap APP in the Golgi and preclude it from the recycling pathway in a manner that reduces β-secretase cleavage and generation of Aβ. Most importantly, the pathogenic relevance of sorLA/LR11 in AD is strongly corroborated in their knockout mouse model: Absence of endogenous sorLA/LR11 showed an elevated level of Aβ, similar to what is observed in AD patients. Further understanding the normal function of sorLA/LR11 in addition to why dramatically decreased amounts of this particular receptor occur in AD patients should provide a framework for designing therapeutic approaches that either prevent the decrease or elevate sorLA/LR11 in hopes of reducing Aβ load. Sorting out the exact mechanisms of intracellular transport and processing in which this recently identified neuronal APP sorting factor participates will surely add to the list of new drug targets aimed at APP-specific therapeutics.
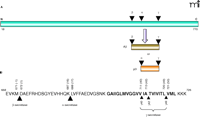
Proteolysis of amyloid precursor protein. A. The membrane-spanning protein amyloid precursor protein (APP) (red) first undergoes ectodomain shedding (i.e., proteolytic cleavage) at either the site of β- or α-secretase activity. This ectodomain shedding process generates the immediate substrate for γ-secretase, which cleaves within the transmembrane domain of APP. Aβ (dark gold) is a product of β- and γ-secretase cleavages. Cleavage by α-secretase (before β-secretase has a chance to cleave APP) results in the peptide termed p3 (dark orange) and precludes the formation of Aβ. The first seventeen amino acids constitute the signal peptide and are not shown. B. Amino-acid residues of Aβ and surrounding region of human APP770 showing the β-, α-, and γ- proteolytic sites. γ-Secretase cleaves at multiple sites within the transmembrane region. Protein sequence is numbered from the first Met. Aβ sequence is indicated in bracket. The transmembrane domain is shown in bold.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Gang Yu, PhD, is an Assistant Professor and the Thomas O. Hicks Scholar in Medical Research at UT Southwestern. His research focuses on the molecular mechanism of Alzheimer Disease. He was responsible for the initial identification of the glycoprotein Nicastrin as a core component of the γ-secretase complex. His lab recently revealed that Nicastrin functions as a receptor for γ-secretase substrates. E-mail: Gang.Yu{at}UTSouthwestern.edu; fax: 214-648-1801.

Sanjiv Shah, BS, currently is pursuing his graduate training in the Center for Basic Neuroscience at UT Southwestern. He works in the lab of Dr. Gang Yu on areas of the γ-secretase complex and substrate recognition. E-mail: Sanjiv.Shah{at}UTSouthwestern.edu.

