The Choline Transporter Resurfaces: New Roles for Synaptic Vesicles?
Abstract
Presynaptic choline uptake is vital to sustained neuronal acetylcholine (ACh) release; however, only with the recent cloning of choline transporters (CHTs) (i.e., SLC5A7), has a picture emerged of the regulatory pathways supporting CHT modulation. Studies arising from the development of CHT-specific antibodies reveal a large, intracellular reserve of CHT proteins, localized to ACh-containing, synaptic vesicles. The intersection of mechanisms supporting vesicular ACh release and choline uptake demonstrates an elegant mechanism for linking regulation of CHT membrane density to rates of ACh release. Furthermore, these studies point to control of the CHT endocytic process as an important target for novel therapeutics that could offset functional deficits in disorders bearing diminished cholinergic tone, including myasthenias and dementias.
Introduction
Acetylcholine (ACh) has widespread actions as a neurotransmitter in the central, peripheral, and autonomic nervous systems, leading to decades of research into strategies to enhance or diminish cholinergic tone for therapeutic ends. Within the mammalian central nervous system (CNS), cholinergic neurons sustain or modulate diverse cognitive and appetitive processes including arousal, attention, memory, and reward (1–3). The addictive properties of nicotine arise from its direct stimulatory effects on central ionotropic ACh receptors, whereas a deficit in cholinergic neurotransmission contributes to the cognitive impairment associated with Alzheimer Disease (AD) (4–6). As degeneration of the basal forebrain cholinergic neurons that project to the cerebral cortex is thought to contribute to dementia in AD (7), enhancement of cholinergic neurotransmission with acetylcholinesterase (AChE) inhibitors is an established, although limited, therapy for managing cognitive decline (6). ACh also serves as a neurotransmitter at the mammalian neuromuscular junction (NMJ) and is essential for neuronal control of muscular contraction (8). Thus, the lethal effects of clostridial neurotoxins arise from a blockade of ACh release at the NMJ located at the diaphragm (9). Here again, reversible AChE inhibitors are clinically beneficial in disorders associated with diminished cholinergic signaling, such as myasthenia gravis. Finally, ACh mediates chemical signaling by preganglionic neurons in both the sympathetic and parasympathetic branches of the autonomic nervous system (10) and is a neurotransmitter at parasympathetic postganglionic synapses, thereby controlling the output of many organs, including the heart, lungs, exocrine and endocrine glands, gut, and bladder (11). Muscarinic receptor agonists and antagonists are used in the treatment of bladder disorders and asthma, respectively; however, the therapeutic potential of current cholinergic drugs is limited by their lack of specificity and the multitude of physiological targets (11).
The presynaptic control of acetylcholine synthesis and release has received less attention as a therapeutic strategy for ACh cholinergic function, despite the fact that the cellular mechanisms controlling ACh synthesis and release at presynaptic terminals are generally well understood and conserved across targets and phyla. At the cellular level, neuronal activity triggers vesicular fusion and the release of ACh into the synaptic cleft. Here, ACh has only a brief opportunity to activate its receptors before being hydrolyzed by AChE to generate choline and acetate (12) (Figure 1⇓). The hydrolysis of ACh terminates cholinergic signaling but the choline is efficiently recaptured by choline transporters (CHTs) to sustain new ACh synthesis. Indeed, pharmacological studies reveal that ACh release cannot be sustained without presynaptic transporter-mediated recapture of choline (10, 13). Once transported, ACh is very efficiently resynthesized by choline acetyltransferase (ChAT) (14, 15), loaded into synaptic vesicles by the vesicular ACh transporter (VAChT) (16–18), and the cycle continues. Acetate might also be recycled at the cholinergic presynaptic terminal (19), although if so, it is by an as yet undefined mechanism. Indeed, glucose metabolism and mitochondrial acetyl-CoA production can provide the needed acetyl moiety for presynaptic ACh homeostasis (20, 21). Regardless, the ChAT Km for choline predicts that this enzyme is not saturated by presynaptic choline levels (22), and that the rate of ACh synthesis saturates in parallel with choline transport at choline concentrations above 5 μ M (23). To enhance ACh production, neurons enhance CHT activity in response to neuronal activity (24), pointing to a mechanism that, if harnessed with novel drugs, could offer an approach to control cholinergic signaling for therapeutic ends. Below, following a review of historical findings that shaped our view of the properties and role of CHT, we describe recently elucidated mechanisms of transporter regulation that depend on the vesicular release process and outline how these findings suggest possibilities for novel molecular interventions to mitigate cholinergic deficits.
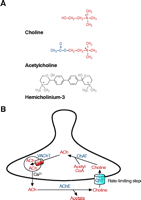
CHT supports presynaptic ACh synthesis. (A) Molecular structures for choline, the substrate of CHT, the neurotransmitter acetylcholine (ACh), and hemicholinium-3 (HC-3), a selective, high-affinity CHT antagonist. (B) At cholinergic presynaptic terminals, CHT functions at the plasma membrane to transport choline into the cytoplasm where it is efficiently acetylated by choline acetyltransferase (ChAT) to generate acetylcholine (ACh). This ACh is packaged into synaptic vesicles by the vesicular acetylcholine transporter (VAChT), a second cholinergic neuron specific transporter. Upon neuron stimulation, ACh-containing vesicles fuse with the plasma membrane in response to elevated cytoplasmic [Ca2+ ]. Within the synaptic cleft, ACh is inactivated by acetylcholinesterase- (AChE)-mediated hydrolysis, and the recapture of choline by CHT is the rate-limiting step in subsequent ACh synthesis.
A Distinct Choline Transporter Supports Synthesis of Acetylcholine
The critical need for presynaptic choline transport in support of ACh production and release first emerged from studies of cholinergic signaling in native preparations such as the perfused cat sympathetic superior cervical ganglia (SCG) (10) and vertebrate NMJ (25, 26). For example, ACh content within the preganglionic terminals innervating the SCG can be maintained even with prolonged stimulation (20 Hz preganglionic stimulation for 60 min) so long as the preparation is bathed in exogenous choline (10). The effect of external choline in sustaining ACh synthesis is saturable in the low micromolar range and can be competitively inhibited by hemicholinium-3 (HC-3) (Figure 1A⇑). HC-3, a bicyclic, constrained choline analog, was initially characterized as a lethal, respiratory paralytic agent (27, 28). These lethal effects of HC-3 [following intraperitoneal (i.p.) injection] were described as similar to the effects of tetanus or botulinum toxins on breathing, but unlike these poisons, HC-3’s effects can be relieved by artificial respiration until the drug is metabolized, revealing its effects to be reversible. Moreover, HC-3 toxicity could be blocked by co-administration of choline, indicating that these two agents likely compete for a common target. The toxic effects of HC-3 treatments are consistent with a cessation of ACh synthesis, failure of neurotransmission at NMJs and subsequent respiratory arrest. Indeed, studies at the NMJ have confirmed that HC-3 suppresses stimulation-evoked ACh release (13, 25). It is unlikely that, following i.p. administration, HC-3’s lethal effect arises from inhibiting the CNS regions that control breathing because of the charged nature of the drug and because of observations that intracerebroventricular delivery of HC-3 depletes ACh levels in the CNS without proving to be fatal (29). The availability of radiolabeled choline of high specific activity allowed for the direct demonstration that HC-3 does indeed act to selectively block presynaptic choline uptake (30, 31) ; sensitivity to low, nanomolar concentrations of HC-3 is now accepted as a major defining characteristic of presynaptic choline uptake (32).
In retrospect, the observations that extracellular choline is required to sustain ACh release and that choline is efficiently recaptured in a saturable, HC-3–sensitive manner provided the essential evidence that cholinergic terminals possess a unique transporter critical for neurotransmitter release. However, the identification of CHT as a molecular entity was thwarted by the low density of mammalian cholinergic terminals in many preparations, relative to more numerous, lower-affinity (but higher-capacity) choline uptake mechanisms found in surrounding cells. Indeed, the relative contribution of cholinergic neuron–specific high-affinity choline uptake to total tissue choline accumulation is small at choline concentrations greater than 10 μM, where the neuronal specific transporter becomes saturated (33, 34). Nonetheless, many important features of the transporter were revealed in metabolic studies. For example, ACh synthesis from exogenous radiolabeled-choline requires extracellular Na+ (33), despite the fact that substantial synaptosomal choline uptake can be achieved at high concentrations in Na+-free buffers. This discrepancy led to the proposal that in cholinergic terminals a distinct high-affinity Na+-dependent choline transporter is dedicated to providing choline for ACh synthesis (23), whereas more widely available choline transport pathways are likely to be present in all cells to support other aspects of choline metabolism, such as phosphatidylcholine synthesis. This idea was confirmed with the identification of two kinetically distinct choline transport activities in rat brain presynaptic terminals: a Na+-dependent, high-affinity (Km = 1–2 μM), low-capacity, HC-3–sensitive (HC-3 Kd = 4–30 nM) (23, 31, 35–39) process linked to cholinergic neurons and a second, low-affinity, higher capacity, Na+-independent uptake process with lower HC-3 sensitivity and ubiquitous distribution (35). As indicated, the majority of the choline transported by the high-affinity, Na+-dependent, HC-3–sensitive mechanism is efficiently converted to ACh (31, 40, 41), suggesting it is both metabolically linked and physically proximal to sites of ACh synthesis. Additionally, choline derived from the AChE-mediated hydrolysis of ACh is transported as efficiently as radiolabeled choline (42), suggesting an as yet unappreciated relationship between AChE and CHT. These findings also indicate that CHT must be highly localized near synaptic ACh release sites, compared to other sites on the presynaptic plasma membrane, to permit efficient recapture of choline.
Other defining characteristics of CHT activity include a requirement for extracellular Cl− and a negative membrane potential, as revealed by inhibition of choline uptake into K+-depolarized synaptosomes (43). We now know that a CHT utilizing similar mechanisms is expressed by all cholinergic neurons (from vertebrates and invertebrates) that have been studied (10, 23, 24, 35, 44–48). Supporting this contention, studies involving the selective destruction (i.e., lesioning) of cholinergic axons––such as those of the septal–hippocampal pathway––eliminate the ability of the target region to support Na+-dependent, HC-3–sensitive, high-affinity choline uptake (49–52). The low-affinity choline uptake mechanism has not been clearly defined at a molecular level but might actually be supported multiple transporters. Among several candidates, we note the activity of the organic cation transporter type 2 (OCT2) that is expressed in neurons and which can transport choline with a Km > 100 μM (53, 54).
A Close Relationship Between Neurotransmitter Release and CHT Regulation
The definition of a cholinergic neuron–specific choline transport process that supports ACh synthesis leads reasonably to questions concerning how the regulation of the CHT parallels changes in ACh synthesis. As noted, when the CHT is functional, ACh synthesis and release are sustained, even during persistent and prolonged stimulation (10, 55). Because ACh itself is not recycled and the transport of choline is rate limiting in the synthesis of ACh, the supply of choline must increase during periods of enhanced ACh release, otherwise the presynaptic vesicle stores of ACh will become depleted. Extracellular concentrations of choline (5–10 μ M) saturate CHT even in the absence of ACh release and hydrolysis; therefore, the only way to enhance CHT-mediated choline uptake is to increase the number of functional CHTs at the presynaptic plasma membrane. Indeed, the maximal rate of CHT-mediated choline uptake (Vmax) is modulated in response to chemical or electrical manipulation of cholinergic firing rates (56–61) (Table 1⇓). For example, in vivo treatments that decrease cholinergic firing and ACh release within the rat hippocampus decrease the Vmax of the CHT, as measured using synaptosomal preparations (56, 57, 62). In contrast, stimuli that enhance in vivo cholinergic firing, ACh release, or both, support an increase in the CHT Vmax (56, 62, 63). The specificity of these in vivo treatments is illustrated by the fact that the general anesthetic pentobarbital, which blunts activity in the septo-hippocampal projection, decreases both hippocampal ACh release and CHT function. Meanwhile, in the striatum where cholinergic activity and ACh release are unaffected by pentobarbital, the CHT Vmax is unaltered. Conversely, scopolamine inhibits presynaptic muscarinic autoreceptors in the hippocampus and striatum and enhances both ACh release and the CHT Vmax in both regions. Although the CHT Vmax is regulated in parallel with the modulation of ACh release in vivo, it is important to note that regulation of CHT can also be demonstrated using nerve terminal preparations in vitro following depolarizing treatments that cause the opening of voltage-gated Ca2+ channels, synaptic vesicle fusion, and ACh release (64–66). These examples provide ample evidence that CHT capacity parallels electrically or pharmacologically induced changes in ACh release. That these changes are physiologically relevant is suggested by behavioral paradigms wherein changes in ACh release also elicit changes in CHT activity (67–71).
Rapid CHT Regulation: A Conserved Relationship with Cholinergic Activation
Tracking CHT With [3 H]-HC-3 Binding
Although HC-3 was initially identified and characterized for its actions as a selective inhibitor of CHT-mediated choline uptake, in its radiolabeled form, the antagonist has also proven valuable as a tool for probing CHT regulation (36, 72) and regional distribution (73, 74). Initially, the utility of HC-3 as a probe for the CHT was supported by findings that the regional abundance of [3 H]-HC-3 binding sites was consistent with the distribution of cholinergic terminals. However, it was soon determined that the antecedent activity of the cholinergic neurons in the preparation also influences the measured density of [3 H]-HC-3 binding sites (75). Indeed, treatments that regulate [3 H]-choline uptake also evoke parallel changes in HC-3 binding (75–78) and thus, the density of [3 H]-HC-3 binding sites (Bmax) appears to closely track the choline uptake capacity of the neuronal membrane. To support such observations, either HC-3 binding sites associated with CHT must become exposed by fusion of intracellular CHT vesicles, or inactive, plasma membrane–resident CHTs lacking high-affinity HC-3 recognition sites are modified to expose new HC-3 binding sites. Many questions remain concerning the apparently simple measurement of HC-3 binding, although new studies, described below, lend support to a vesicular origin of the new HC-3 binding sites.
Molecular Cloning and Characterization of CHT
Although more than four decades of CHT characterization ensued from its original definition, only recently have the genes that encode CHT proteins been identified. Initial evidence supporting the existence of specific CHT-encoding genes was obtained by eliciting the expression of HC-3–sensitive choline uptake in Xenopus laevis oocytes following injection of rat spinal cord mRNAs (79). Elsewhere, purification and reconstitution efforts targeted CHT activity in cholinergic neuron–rich insect ganglia (47, 80). A false lead to the identification of CHT, perhaps triggered by the dual dependence of choline transport on both extracellular Na+ and Cl−, placed CHT as a member of the Na+/Cl− neurotransmitter-transporter gene family that also contains the transporters for γ -amino butyric acid (GABA), serotonin, and dopamine (81). The gene in question turned out, however, to encode a creatine transporter (82). Unfortunately, many databases still refer to the creatine transporter as a choline transporter, or CHOT1. An ingenious yeast expression cloning strategy also identified a choline transporter, now termed CTL1, in a search for the missing CHT (83). However, its ion-dependence, kinetic and pharmacological sensitivities, and regional distribution did not fit with expectations regarding presynaptic choline uptake. A major breakthrough in this saga was achieved with the discovery of a novel transporter-like protein exhibiting cholinergic neuron specific expression in the nematode Caenorhabditis elegans (84). When expressed in X. laevis oocytes, the corresponding C. elegans cRNA (now known as CHO-1), as well as a homologous rat CHT cRNA, were able to confer HC-3–sensitive, high-affinity, Na+ - and Cl− -dependent choline transport activity. These observations paved the way for the identification of CHT cDNAs and genes in multiple species, including mouse and human (84–88). Initial cloning reports used the name CHT1 to describe this gene product in humans (87, 89), rats (84), and mice (86). Since that time, analysis of genome sequencing results predicts the presence of only a single CHT gene in multiple species. For example, in addition to the characterized human (NM_021815), rat (NM_053821), mouse (NM_022025), Torpedo marmorata (AJ420808) and C. elegans CHT (NM_070138) orthologs, a single CHT-encoding gene sequence is predicted in the Drosophila melanogaster (NM_142486) and Anopheles gambiae (XM_312589) genomes. Although initial caution concerning the possibility of multiple CHT genes and isoforms led to the term CHT1, in the absence of any additional predicted CHTs, we have adopted the simple “CHT” designation, leaving additional annotation as needed for possible mRNA processing variants.
CHT is a member of the SLC5 family of mammalian Na+ -dependent transporters (in humans, CHT is designated as SLC5A7) that currently consists of eight members (90) and that is part of a larger sodium-solute transporter superfamily, with several hundred members identified in species from humans to bacteria (91–93). Within the SLC5 family, the best-characterized members are the Na+ -dependent glucose transporters (SGLTs), which participate in the intestinal and renal absorption of water and glucose (94, 95), and the Na+/I− symporter (NIS), responsible for iodide uptake in the thyroid gland (96). CHT is the only member of this family with a known presynaptic function related to neurotransmitter synthesis, although another family member (SLC5A4) has been proposed to serve as a glucose sensor in cholinergic motor neurons (97) and could contribute, in an as yet not understood role, to the control of cholinergic neurotransmission.
The CHT genes identified in mice, rats, and humans reside on chromosomes 17, 9, and 2, respectively (87). The structure of mammalian CHT genes is well conserved: each has nine exons and spans ~25 kb of genomic sequence (Figure 2A⇓) (84, 86, 87, 89). Sequence similarity between CHT and other SLC5A family members, as well as hydrophilicity analyses and topology prediction algorithms, has led to the revision of initial predictions of twelve transmembrane segments to a new prediction comprising thirteen transmembrane domains with an extracellular N terminus and a cytoplasmic C terminus (87, 91) (Figure 2B and C⇓). A consensus site for N -linked glycosylation is present at residue N301 in the fourth extracellular loop of human, rat, and mouse CHTs, a prediction confirmed through immunoblots from COS-7 cells transfected with either wild-type human CHT or a CHT-N301Q mutant (Figure 2D⇓). Using electron microscopy (EM) immunogold labeling techniques, we have demonstrated that the CHT C terminus resides in the cytoplasm (66) (see also Figure 3D and E⇓). Although this initial model offers an important framework to guide structure-based investigations of CHT regulation, further analysis into the details of CHT transmembrane topology is warranted. With respect to predictions of the CHT quaternary structure, it is worth noting that freeze-fracture analysis of SGLT1 indicates that this SLC5A family member exists as a monomer on the plasma membrane (98).
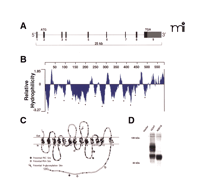
CHT revealed. (A) The hCHT gene structure comprises 9 exons encoding a 5 kb transcript distributed over a span of ~25 kb on chromosome 2. (B) Hydrophilicity analysis of the 580 amino-acid protein encoded by the 1740 bp coding region of hCHT predicts 13 transmembrane-spanning domains (TMDs, marked by asterisks). (C) The predicted transmembrane topology for hCHT includes an extracellular N-terminus and an intracellular C terminus. Amino acids conserved from humans through mice and rats to C. elegans are depicted with filled circles. There is a single predicted extracellular N-linked glycosylation site (N301) and multiple cytoplasmic protein kinase A and protein kinase C consensus phosphorylation sites [Reproduced with permission from (87) ]. (D) The predicted N-linked glycosylation site was converted to glutamine (N301Q mutant) by site directed mutagenesis and the resulting construct was transfected into COS-7 cells. Compared to the wild-type hCHT, immunoblot analysis of this N301Q mutant reveals a loss of the major glycosylated form of hCHT and an increase in the abundance of nonglycosylated CHT. Similar shifts in mobility have previously been observed following Peptide N-Glycosidase F (PNGase F) treatment of hCHT (66).
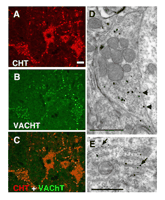
CHT colocalizes with VAChT at presynaptic terminals of cholinergic neurons. (A) In the ventral horn of the mouse spinal cord, CHT immunoreactivity is present in motor neuron cell bodies as well in the large presynaptic C-boutons that surround them (Scale bar = 20 μ m). (B) VAChT is present in the same cell bodies and terminals as CHT, confirming their cholinergic identity. (C) The distributions of CHT and VAChT overlap completely demonstrating that CHT is a constant feature of cholinergic neurons and that it is enriched at presynaptic sites. (D) Immuno-electron microscopy with CHT-specific antibodies illustrates that within an individual cholinergic presynaptic C-bouton, CHT immunoreactivity revealed by silver enhanced gold particles is predominantly associated with presynaptic vesicles and is only occasionally detected at the plasma membrane (arrows). Note that the epitope is on the cytosolic face of the plasma membrane, consistent with the predicted topology of CHT (scale bar = 1 μ m). (E) In the cell body of a cholinergic neuron, a biosynthetic pool of CHT immunoreactivity is labeled with the C-terminal epitope found on the cytoplasmic face of the rough endoplasmic reticulum (scale bar = 1 μ m). [Data adapted from (66).]
Importantly, the proposed topology model indicates potential serine and threonine phosphorylation sites in the fifth intracellular loop and C terminus that could serve to regulate CHT function (87). Gates et al. have demonstrated that CHT is indeed a phosphoprotein; the phosphorylation of synaptosomal CHT is triggered by kinase activators, such as phorbol 12-myristate 13-acetate (PMA) (99). It seems likely that CHT activity and/or distribution is influenced by interacting proteins, and perhaps CHT complexes with kinases or phosphatases might serve to regulate CHT in a manner analogous to the relationship between the presynaptic monoamine transporters and protein phosphatase 2A (100). Indeed, CHT function is diminished in response to inhibitors of protein phosphatases including okadaic acid, calyculin A, and cyclosporine A (88, 99, 101), suggesting that CHT phosphorylation may be linked to regulation of the abundance of active CHTs at the plasma membrane.
Observing CHT at the Synapse
As noted, the first direct visualization of CHT distribution was accomplished using [3 H]-HC-3 autoradiography (73, 74, 102–107). As with choline uptake, the specificity of [3 H]-HC-3 autoradiographic labeling of cholinergic terminals could be validated through the specific lesioning of cholinergic pathways (52). Although autoradiography is a powerful approach—for example, in offering the opportunity to discern the fine distribution of CHTs in distinct brain subregions (108) —no synaptic-level resolution is provided, and substantial evidence indicates that HC-3 binding to CHT is subject to regulation in response to changes in cholinergic activation (75). Because the abundance of HC-3 binding sites likely reflects a combination of CHT availability and the relative activity of cholinergic neurons, problems in interpretation of drug- or disease-induced changes arise. For example, such issues may confound the interpretation of [3 H]-HC-3 binding data to measure changes in the density of cholinergic terminals in AD as both decreases (105, 109) and increases (110, 111) in CHT density have been reported in postmortem samples. These discrepant findings could reflect an overall decrease in the number of cholinergic terminals (7) in AD, balanced in a region- or age-dependent manner by compensatory counter regulation of CHT in the remaining cholinergic axons. The limited resolution of autoradiographic studies also precludes an understanding of the fine localization of CHTs, such as might support the efficient recapture of synaptic choline released after AChE-catalyzed hydrolysis.
The development of CHT-specific antibodies has led to the direct, activity-independent detection of CHTs (66, 112–115). These studies, employing multiple CHT-specific antibodies raised against distinct epitopes, have confirmed a distribution of CHT protein in register with the localization and projections of cholinergic neurons. Consistent with its presumed primary role in support of ACh synthesis, CHT immunoreactivity is concentrated at presynaptic sites where it colocalizes with VAChT (Figure 3⇑). There is no evidence at present to indicate the presence of CHT immunoreactivity in noncholinergic neurons or glia, although expression of CHT outside the nervous system has been reported (116, 117). With CHT-specific antibodies available that can reveal sites of transporter expression in the human CNS (66, 112), new opportunities are evident to evaluate CHT abundance relative to other cholinergic markers in AD and other disorders associated with cholinergic dysfunction.
Hidden Pools of Vesicular CHT
Although CHT immunoreactivity is present both in the cell bodies and presynaptic terminals of cholinergic neurons (66, 112, 113), choline uptake activity is restricted to the presynaptic terminals (118, 119). We used immuno-EM analysis to define CHT subcellular localization and identified several distinct pools of CHTs within cholinergic neurons (66). At the cell body level, abundant CHT immunoreactivity is evident in association with the rough endoplasmic reticulum membranes (Figure 3E⇑). This biosynthetic pool likely accounts for the CHT signal associated with cholinergic soma, because CHT immunoreactivity is not abundant at the cell soma plasmalemma. In contrast, CHT immunoreactivity is evident at the plasma membrane of presynaptic terminals, although by far the greatest density of CHT labeling is surprisingly found to be associated with intracellular vesicles (Figure 3D⇑). The possibility exists that epitope masking may limit the visualization of pools of plasma membrane–localized CHT, although support for a chiefly vesicular localization was also obtained using biochemical fractionation and vesicle immunoisolation techniques (66). Importantly, CHT-positive vesicles were found to harbor multiple synaptic vesicle proteins and to contain ACh. Because CHT requires a Na+ gradient to drive choline transport, it seems unlikely that CHT functions on the vesicle membrane as a choline importer. More likely, as we discuss below, a vesicular localization is one step in a multistep process that allows linkage of vesicular fusion, ACh release, and activity-dependent increases in choline transport, localized to the synapse.
The mechanisms supporting sorting and trafficking of CHT to nerve terminals are unknown but may involve directed axonal export of CHT vesicles or the retrieval of axonal proteins following constitutive sorting to dendrites and their subsequent retention at presynaptic sites (120, 121). Furthermore, distinct subtypes of vesicles and molecular motors serve to deliver scaffolding and synaptic vesicle components to presynaptic terminals (122–124). As CHT and VAChT both come to reside on common synaptic vesicles at the nerve terminal, it is possible that they share common targeting signals from an early stage to allow for their proper and coordinated trafficking. Additionally, we speculate that CHT and VAChT may share common structural motifs for endocytic vesicle targeting. For example, the internalization and sorting of VAChT to the synaptic-like microvesicles (SLMVs) in PC12 cells is dependent on an acidic residue–flanked dileucine motif in its cytoplasmic C terminus (125, 126). A similar motif is apparent in the cytoplasmic C terminus of mammalian CHTs (I529, L530) that warrants investigation with respect to a role in the endocytosis and vesicular sorting of CHT. The abundance of CHT at the plasma membrane and the uptake of choline into CHT-transfected cells are greatly enhanced (Figure 4⇓) when clathrin-mediated endocytosis is blocked by cotransfection of a dominant-negative mutant of dynamin I (127). Enhanced choline transport has also been reported in CHT-transfected cells in response to disruption of endocytosis with a C-terminal fragment of AP180 (128). These studies illustrate how blocking the endocytosis of a constitutively recycling CHT protein results in its accumulation on the plasma membrane and indicate that efficient CHT endocytosis may serve both to target CHT to synaptic vesicles and negatively regulate choline uptake capacity. With an improved understanding of the mechanisms recruiting CHT for internalization, the development of selective pharmacological inhibitors of CHT endocytosis could provide a means to therapeutically enhance cholinergic function.
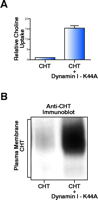
CHT accumulates on the plasma membrane when constitutive endocytosis is impaired. (A) We have observed that cotransfection of COS-7 cells with hCHT plus the dominant negative K44A mutant of dynamin I (127) elicits 15.6 +/– 1.1 fold more HC-3 sensitive [3 H]-choline uptake than cotransfection of hCHT with an empty vector control. (All assays performed in triplicate, n = 3, p < 0.0005.) (B) Cell surface biotinylation confirms that hCHT accumulates on the plasma membrane when clathrin-mediated endocytosis is blocked by expression of the K44A mutant of dynamin I.
The CHT Resurfaces
Above, we reviewed the close relationship between the rate of ACh release by cholinergic neurons and their capacity for choline uptake. These observations could reflect a direct mechanism for coupling ACh release to choline reuptake or, alternatively, reflect parallel but distinct pathways linked to neuronal activity. Our most recent evidence provides confirmation that CHT plasma membrane density is augmented by stimuli that trigger ACh release (66). We found that depolarization-evoked increases in synaptosomal CHT Vmax can be blocked by the pretreatment of hippocampal synaptosomes with botulinum neurotoxin C, an agent known to inhibit the vesicular release of neurotransmitter by proteolysis of syntaxin 1A (129). Moreover, we found that elevations in transport activity require extracellular Ca2+ and can be blocked by voltage-dependent Ca2+ channel antagonists, consistent with prior observations (64). Finally, we observed a significant increase in the amount of CHT protein located at the synaptic membrane following depolarization. Together, these observations lead to a new model for acute CHT regulation (Figure 5⇓), whereby CHT is delivered to the plasma membrane by a fraction of the same synaptic vesicles that mediate ACh release. Conversely, the presence of CHT on synaptic vesicles indicates that it must also be endocytosed from the plasma membrane in conjunction with other synaptic vesicle proteins, and the rate of recruitment of CHT to clathrin-coated pits will help dictate the extent of membrane choline uptake capacity achieved.
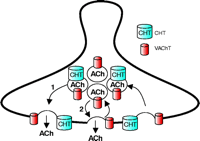
Localization of CHT to a subset of synaptic vesicles couples plasma membrane delivery of CHT to the rate of ACh release. Although CHT functions at the plasma membrane to capture choline for ACh synthesis, recent results indicate that it is also abundantly localized to a subset of VAChT-positive, ACh-containing synaptic vesicles (66). Thus, ACh release occurs by the fusion of two distinct but equally abundant subsets of synaptic vesicles, one that contains both CHT and VAChT and a second class of vesicles containing only VAChT. At steady state in presynaptic terminals, no major pools of CHT exist on VAChT-negative vesicles; therefore, ACh release and CHT insertion into the plasma membrane are very intimately linked.
An important question regarding the coordination of CHT and VAChT sorting arises from our identification of heterogeneity within the populations of CHT- and VAChT-containing synaptic vesicles. In mouse striatum, CHT-positive synaptic vesicles represent approximately 50% of the VAChT-positive synaptic vesicles. This does not arise from differential cellular expression because both proteins are present at all cholinergic terminals (Figure 3⇑). Rather, this observation could represent either a random distribution of less highly expressed CHT among the total population of cholinergic, VAChT-positive, synaptic vesicles or the presence of specific targeting information that confines CHT to a specific subpopulation of cholinergic synaptic vesicles. Conceivably, there may exist a rapidly recycling pool of VAChT-only vesicles whose fusion dynamics are controlled independently from the CHT–VAChT pool of vesicles. In this regard, synaptosomal choline uptake and activity-dependent upregulation of CHT are inhibited by microtubule depolymerizing agents such as colchicine (130) and vinblastine (65), whereas ACh release seems less significantly affected (130, 131). If two pools of vesicles provide nonredundant aspects of cholinergic signaling, it may be possible to identify agents that can selectively stabilize fusion of one over the other, mobilizing, for example, the CHT–VAChT pool to achieve an increase in presynaptic choline uptake and enhanced ACh production.
Is Trafficking All There is to CHT Regulation?
The large reserve of CHTs on cholinergic synaptic vesicles and the Ca2+ -dependent, botulinum neurotoxin C–sensitive regulation of choline uptake indicates a role for trafficking in coupling CHT regulation to ACh release (66). However, previous investigations of CHT regulation have arrived at models that account for CHT regulation based on the activation of previously silent plasma membrane CHTs (132). Therefore, it is important to critically reevaluate these earlier conclusions with the hindsight gained from the more recent investigations to integrate results into a coherent model that can guide future experiments. As noted, although at steady state most CHTs reside on synaptic vesicles, relative rates of CHT exocytosis and endocytosis and the half-life of CHT at the plasma membrane have not yet been carefully quantified. Depolarization of synaptosomes with high [K+ ] buffers evokes immediate increases in ACh release, whereas persistent changes in CHT function only become maximal after more than ten minutes of stimulation (64, 65, 76). However, once the rate of choline uptake has been modulated, the effect persists for many minutes following removal of the depolarizing stimulus. These temporal discrepancies between CHT regulation and ACh release suggest that in addition to trafficking, maximal CHT function may require further mechanisms to either activate or stabilize the transporter at the plasma membrane (Figure 6⇓).
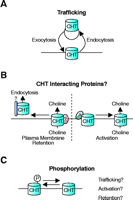
Potential mechanisms for the regulation of CHT function. Each of these mechanisms represents a candidate target for the pharmacological enhancement of CHT function. (A) A trafficking component is supported by the large cytoplasmic reserve of CHT on cholinergic synaptic vesicles that could serve to couple choline uptake capacity to the rate of ACh release (66). CHT abundance at the plasma membrane will reflect the balance between CHT exocytosis and endocytosis. (B) There are many possible ways to regulate CHT through interacting proteins. Different binding partners could either retain CHT at the plasma membrane or lead to CHT’s recruitment into clathrin-coated pits for endocytosis. Alternatively, the activity of CHTs within the plasma membrane could be regulated by interactions between CHT and other as yet unidentified proteins. (C) In conjunction with trafficking mechanisms and protein–protein interactions for controlling CHT function, posttranslational modification of the transporter could also serve control presynaptic choline uptake. The CHT phosphorylation state could directly regulate transport activity but might also determine the affinity of interactions between CHT and various regulatory or trafficking proteins.
Modulation of CHT activity resident in the plasma membrane could occur in response to posttranslational modifications such as phosphorylation or ubiquitination or through the interaction with regulatory proteins. Separate regulated steps of plasma membrane insertion followed by transporter activation have been elucidated for other plasma membrane transporters, including the insulin-regulated glucose transporter of muscles and adipocytes (GLUT4) (133), Na+ -K+ -Cl− cotransporters in epithelial cells (134), as well as the presynaptic norepinephrine transporter (135). From a technical point of view, directly monitoring changes in transporter activity is currently difficult but can be inferred from data demonstrating changes in transporter function that are not matched by equivalent changes in plasma membrane abundance of the transporter.
An example of CHT regulation that is potentially trafficking-independent involves changes in HC-3 binding triggered by arachidonic acid. The treatment of rat striatal plasma membrane–enriched homogenates with phospholipase A2 (PLA2) or its product, arachidonic acid, results in a twofold increase in the Bmax for [3 H]-HC-3 binding (132, 136, 137). If this membrane preparation is depleted of CHT-containing vesicles, then trafficking would be precluded and the reported appearance of new HC-3 binding sites must represent the activation of previously silent plasma membrane CHTs. However, the depletion of vesicular CHT from this preparation has not been established and will require more direct confirmation. Regardless, other evidence solidifies a role for PLA2 activity in the regulation of CHT. For instance, the effects of depolarizing pretreatments on choline uptake into both mammalian and invertebrate cholinergic neurons can be blocked by quinacrine (132) or arachidonyltrifluoromethyl ketone (138), both PLA2 inhibitors. Cytoplasmic PLA2 activity is calmodulin (CaM)-dependent; thus, reports of the blockade by CaM inhibitors of depolarization-stimulated increases in CHT function are consistent with a role for PLA2 in this process (139). Each of these examples supports an important role for PLA2 activation in the regulation of CHT in response to neuronal activity, but the exact step regulated by arachidonic acid remains elusive. Although the concept of a post-insertion, arachidonic acid–dependent CHT activation is attractive, other explanations for these results are also plausible. Treating presynaptic plasma membranes with arachidonic acid can promote the fusion of synaptic vesicles (140, 141), which could also expose previously occluded HC-3 binding sites within the lumen of vesicles. Furthermore, because quinacrine can block depolarization-stimulated ACh release from synaptosomes (142), it is premature to conclude that quinacrine also blocks a subsequent trafficking-independent step in CHT modulation. Finally, with intact striatal slices or synaptosomes, the effects of arachidonic acid are not additive with the enhancement of HC-3 binding evoked by elevated K+ –induced depolarization (132). These observations suggest that a common mechanism is utilized for CHT enhancement linked to the mobilization of vesicular reserve pools or subsequent CHT membrane stabilization.
In most studies, there is a strong correlation between regulation of HC-3 binding and choline transport. However, observations that the affinity for HC-3 membrane binding can be shifted between two distinct affinity states in a Mg2+ - and ATP-dependent manner has further complicated the interpretation of HC-3 binding (38, 143). Only one of these two HC-3 affinity states is predicted to correspond to the active form of CHT that transports choline (38). Another example of divergence between regulation of choline uptake and HC-3 binding is the chelerythrine-induced stimulation of CHT-mediated choline uptake into horseshoe crab (Limulus polyphemus) brain slices that is not paralleled by an increase in HC-3 binding (144). Although the targets of chelerythrine in Limulus have not been identified—mammalian protein kinase C isoforms are sensitive to this drug (145) —the separable effects of this treatment on choline uptake and HC-3 binding are intriguing as they support the presence of a trafficking-independent form of CHT regulation. Interestingly, a coding SNP in the human CHT that results in an I89V substitution in transmembrane domain 3 has been reported to reduce the Vmax for choline uptake without affecting whole-cell HC-3 binding (146), again suggesting that choline uptake and HC-3 binding by CHT are separable processes. Further studies taking advantage of specific antibodies, surface biotinylation protocols, and mutant CHT cDNAs are needed to advance our understanding of how CHT regulation is connected to changes in HC-3 binding.
Adding Mustard to the Mix
Hemicholinium mustard (HCM) is a derivative of HC-3 that can covalently modify and irreversibly inactive CHTs on the plasma membrane of living neurons (147). The rate of recovery of choline uptake in HCM-treated cholinergic neurons (in horseshoe crab brain-slice preparations) is acutely enhanced by treatments that cause depolarization (144). This finding supports the presence of a reserve pool of HCM-insensitive CHTs tucked within these neurons whose exposure is modulated by depolarization. However, because surface-expressed CHT possessing a very low affinity HC-3 binding site would not likely bind HCM either, these studies cannot unequivocally rule out the presence of silent transporters residing in the plasma membrane awaiting activation. Interestingly, choline mustard also selectively inactivates CHT in an irreversible manner but, in addition, is a substrate for the transporter (148). Unlike HCM, choline mustard pretreatment of synaptosomes effectively blocks subsequent depolarization-induced increases in choline uptake (149). As choline mustard is transported into the presynaptic cytoplasm, it can conceivably exert its effects by alkylating both plasma membrane as well as vesicular CHTs. Taken together, these studies are compatible with a trafficking explanation for CHT regulation. However, this interpretation is also limited by uncertainty concerning the nature of the HC-3 binding sites (and whether choline mustard targets the same sites), and it remains possible that interconversion of plasma membrane pools of CHT accompanies transporter activation.
Animal Models for Studies of CHT Regulation
Transgenic mice overexpressing AChE exhibit increased choline uptake and HC-3 binding in the hippocampus (150, 151). Furthermore, in vivo microdialysis revealed that hippocampal extracellular ACh levels are comparable between AChE transgenic mice and wild-type controls, suggesting that the increases in CHT function might have helped to normalize cholinergic function in these mice (151). Interestingly, CHT protein levels are also increased in the striatum of AChE knockout mice (152), whereas VAChT expression and ChAT activity remained unchanged. The apparent increases in CHT function in both the AChE knockout or transgenic mice suggest that multiple steps ranging from CHT transcription to CHT trafficking, posttranslational regulation, and degradation likely conspire via CHT to control presynaptic choline availability. Altered signaling through muscarinic ACh receptors is likely to be involved in some of the changes in CHT function in response to AChE manipulations. Knockout mice for the m2 and m4 muscarinic receptors have been generated and characterized with respect to the roles of these receptors in mediating negative feedback on presynaptic ACh release (153). In light of the potential role of synaptic vesicles in the delivery of CHT to the plasma membrane, diminished inhibition of ACh release in autoreceptor mutant mice could also increase CHT abundance at the plasma membrane. Of course, presynaptic muscarinic receptors are also logical candidates for communicating altered ACh release rates to plasmalemma CHTs via possible activation mechanisms independent of trafficking as well. One additional model that might provide information about CHT regulation is the ChAT hemizygous mouse. Whereas homozygous disruption of ChAT expression is lethal (8, 154), ChAT hemizygotes are viable and fertile. The loss of a single ChAT allele represents a cholinergic neuron-specific genetic alteration that could serve as a model for testing whether functional compensation targets CHT mobilization or activation to maintain stores of presynaptic ACh.
The mouse CHT gene maps to chromosome 17C, a region that has been associated with differences in high-affinity choline uptake by quantitative trait loci analysis (155). The mapping of variability in CHT function to the vicinity of the CHT gene indicates that these results could reflect functional polymorphisms in the CHT coding region or within the CHT promoter. However, until these loci are definitively mapped to the CHT gene, it remains possible that the effects on choline uptake have arisen from other alterations in cholinergic neuron function. A separate study indicates the presence of a nonsynonymous single nucleotide polymorphism (SNP) in the murine CHT gene (156) ; however, the impact of this or other polymorphisms on CHT function remains untested. We have recently developed CHT knockout mice (157), and our initial findings indicate that these mice die within an hour of birth, with symptoms consistent with paralysis and a loss of respiratory function, as expected for deficits in cholinergic neurotransmission at the NMJ and similar to the phenotype of the ChAT knockouts (8, 154). Interestingly, the CHT hemizygotes are viable and healthy and appear to have compensated for loss of a single CHT allele through posttranslational processes. Clearly, these mice offer many opportunities to illuminate endogenous mechanisms of CHT regulation and could well serve as a model to identify potential human phenotypes associated with carriers of CHT loss-of-function alleles.
CHT and Human Disease
No diseases with obvious cholinergic implications have been associated with the hCHT locus at chromosome 2q12 (87). Nonetheless, CHT and ChAT function in a common pathway, and the physical characteristics of mice harboring knockouts for each gene singly appear similar; therefore, mutations of either of these genes in humans might have similar consequences. Mutations in ChAT elicit congenital myasthenic syndrome associated with episodic apnea (CMS-EA) (158, 159). Possibly, other cases of idiopathic myasthenic syndromes arise from a genetically determined loss of CHT expression or activity and a subsequent deficit in ACh synthesis. The presence of a relatively common nonsynonymous hCHT SNP (I89V) that yields diminished choline uptake activity in transfected cells (146) certainly raises intriguing questions as to whether CHT may be an important risk gene for disorders bearing deficits in cholinergic function. AD patients represent one candidate population that might be vulnerable to deficits in CHT expression or regulation: a loss of nucleus basalis cholinergic neurons occurs in the brains of AD patients (7), and the symptoms of dementia can be relieved with AChE inhibitors (6). Even moderately diminished CHT-mediated choline uptake might act synergistically on the sensitized background of a loss of cholinergic neurons to exacerbate functional cholinergic deficits. The reported increase of HC-3 binding in the cerebral cortex of AD brains suggests that CHT function may undergo compensatory upregulation in order to bolster the effectiveness of the surviving cholinergic neurons (110, 111, 160). Future efforts to target drugs that can enhance CHT function would thus appear to be desirable. Rather than developing CHT antagonists, small molecules that could bind to CHT or a CHT regulatory factor and block transporter endocytosis might be ideal. Although this seems highly ambitious, some progress has already been made in developing such choline uptake enhancers, (MKC-231, for example) (161). This agent reportedly enhances HC-3-sensitive choline uptake and reverses learning deficits in a rat model of hypocholinergic function (162). It is unknown whether MKC-231 targets CHT directly or whether it causes more global activation of cholinergic neurons. With the availability of CHT antibodies and additional insights arising from subcellular studies, an answer should now be readily attainable. Furthermore, the availability of CHT expressed in model cell systems should support high-throughput screens to identify agents eliciting enhanced choline uptake independent of changes in cholinergic neuronal activation. By targeting pathways likely to participate in posttranslational control of CHT trafficking, we have succeeded in enhancing choline uptake in model systems by as much as 15-fold (Figure 4⇑). Studies are now planned to expand this effort to chemical libraries and to ascertain whether choline uptake enhancement exerts a meaningful impact on ACh synthesis and release.
Conclusions
CHT plays an important role in supporting and regulating presynaptic ACh synthesis. However, focused studies on the cell biology and pharmacology of the CHT protein have long been limited by the challenges surrounding the identification of the CHT gene. The recent cloning of CHTs from multiple species and advances in our understanding of CHT’s subcellular localization have revealed a large reserve of CHTs on synaptic vesicles, providing an elegant mechanism to link presynaptic choline uptake to antecedent levels of neuronal activity. Although, to date, the only drugs available to target CHT have been blockers whose effects are lethal, the stage is now set to identify positive modulators of CHT that could provide relief from cholinergic deficiency. Very likely, strategies to understand how CHT enhancement can be achieved will follow from an increasingly sophisticated understanding of how CHT localization and activity are coordinated in healthy cholinergic neurons.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Shawn M. Ferguson, BS, is an advanced graduate student in the Neuroscience Graduate Program at Vanderbilt University and is performing thesis research in the Blakely laboratory.

Randy D. Blakely, PhD, is the Allan D. Bass Professor in the Department of Pharmacology and Director of the Center for Molecular Neuroscience at Vanderbilt University. Address correspondence to RDB. Email: randy.blakely{at}vanderbilt.edu; fax (615) 936-3040.

