Looking at Lithium: Molecular Moods and Complex Behaviour
Abstract
Lithium and other mood-stabilizing drugs are used for the management of bipolar mood disorders and, to a lesser extent, for augmentation of other psychoactive drugs. Lithium also has neuroprotective properties that may be useful for treatment of neurodegenerative diseases such as Alzheimer’s disease and amyotrophic lateral sclerosis. Over the years, lithium has been shown to inhibit inositol monophosphatases and glycogen synthase kinase 3, but the relevance of such enzyme inhibition to the therapeutic effects of lithium has remained difficult to assess. Here, we provide an overview of recent advances in the identification of molecular mechanisms involved in the regulation of behavior by lithium. We also highlight recent findings suggesting that lithium could exert some of its behavioral effects by acting on a dopamine receptor regulated signaling complex composed of Akt, protein phosphatase 2A, and the multifunctional protein scaffold beta-arrestin 2.

Since its approval in 1970 for the treatment of bipolar illness, lithium has become a commonly prescribed mood stabilizer, with use in depression and psychotic disorders and potential use in neurodegenerative disorders such as Alzheimer disease. The predominant explanation for lithium’s actions (e.g., in FDA labeling) was for many years limited to alterations in sodium transport in excitable cells and a “shift” in catecholamine metabolism. Remarkably, specific enzyme activities were subsequently identified as targets of the simple cation, and the roles of these and other proteins in psychiatric disease were corroborated as genomic data and animal models of mood became available. New insights into catecholamine signaling, concerning the specificities of receptor subtypes and novel modalities of receptor signaling, continue to broaden our understanding of lithium’s actions. In some instances, lithium may affect mood-regulating enzymes that normally utilize magnesium as a cofactor; protein–protein interactions that control neuronal receptor pathways also appear to be subject to disruption by lithium. These insights have implications not only for the development of new psychotropic medicines, but also for our understanding of mood and behavior as functions of discrete molecular interactions.
Introduction
Since the discovery of its therapeutic effects on mania (1, 2) lithium has become the prototypical member of a small family of structurally disparate mood stabilizers. These drugs are used primarily for the management of bipolar disorders and cyclothymia—two psychiatric ailments, each affecting one to two percent of the US population, characterized by alternating episodes of manic and depressive symptoms. In addition to bipolar disorders, lithium is also used as part of combination therapies with drugs affecting monoaminergic neurotransmission, such as antidepressants and antipsychotics (3, 4). Clinical trials have shown that lithium in combination therapy can be effective for treatment-resistant forms of depression (5). Furthermore, lithium has also been reported to lower suicide rates in individuals with bipolar disorder, major depression, or schizoaffective disorders (6, 7).
Apart from its effects on psychiatric illnesses, recent findings have suggested that lithium could also have, at least in preclinical animal models, beneficial effects in several neurodegenerative conditions. Lithium has been shown to reduce the toxicity of mutant Huntington disease protein in flies (8) and improve behavioral and histological phenotypes in a transgenic mouse model of spinocerebral ataxia (9). Lithium also reduces the phosphorylation of the microtubule associated protein tau (10)—an important component of neurofibrillary tangles—and the production of beta-amyloid peptides (11) in transgenic mouse models of Alzheimer disease. Finally, lithium is neuroprotective in mice that overexpress a mutant form of superoxide dismutase 1 (SOD1) that is associated with amyotrophic lateral sclerosis (ALS), and it is also neuroprotective in patients with ALS (12–14).
Despite important clinical applications, the molecular mechanisms by which lithium and other mood stabilizers exert their therapeutic effects in humans or preclinical animal models are not well understood. Over the years, lithium has been shown to inhibit various enzymes, including inositol monophosphatases (IMPAs) (15) and glycogen synthase kinase 3 (GSK3) (16, 17). However, in the absence of clear etiological animal models for bipolar disorders, the implications of these targets in the therapeutic actions of lithium have remained less than fully explored.
For a molecule or group of molecules to be established as direct targets of lithium in the regulation of behavior, a number of criteria should be satisfied: 1) The “target” must be directly and specifically affected by lithium in vitro; 2) the target must be affected by lithium in the mammalian brain (at serum concentrations of 0.6 to 1.5 mM); 3) activation or inhibition of the target should recapitulate the effects of lithium in vivo with respect to established biochemical markers; 4) pharmacological and genetic modulation of target activity should reflect the effects of lithium in model systems of depression or mania; and 5) elimination of the target should abolish both biochemical and behavioral responsiveness to lithium in these models.
In this article, we provide an overview of recent advances in the identification of molecular mechanisms involved in the regulation of behavior by lithium. We limit our review of targets affected by lithium, in vitro or in vivo, to studies that have addressed behavioral components of the potential targets. We also highlight recent findings suggesting that lithium may exert some of its effects not by directly inhibiting enzymes, but by destabilizing specific protein complexes involved in the regulation of enzyme activity. One such complex, identified in the brain through the activation of dopamine D2 receptors, is composed of protein kinase B (Akt), protein phosphatase 2A (PP2A), and the multi-functional protein scaffold β-arrestin 2 (βArr2) (18). This principle may have relevance to the previously proposed targets of lithium.
The Inositol Depletion Hypothesis
One of the first compelling mechanisms proposed for lithium action arose from its characterization as an uncompetitive inhibitor of various phophoinositol phosphatases, with kinetic parameters compatible with therapeutic concentrations (15). Inositol derivatives are one of the major second messenger systems in cells (Figure 1⇓). Activation of phosphoinositide-specific phospholipase C (PLC) isoforms by G protein–coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs) leads to the cleavage of membrane phosphatidylinositol bisphosphate (PIP2) into dyacylclycerol (DAG) and inositol-1,4,5 trisphosphate (IP3) (15, 19). DAG activates protein kinase C (PKC) isoforms, whereas IP3 triggers the release of calcium from intracellular stores and thus leads to activation of cacium-regulated signaling proteins such as calcineurin (PP2B) and calcium calmodulin regulated kinases (CaMKs). Subsequently, the second messenger IP3 is rapidly hydrolyzed, by an enzymatic cascade involving multiple phosphoinositol phosphatases, to terminate the signal and recycle inositol for the crucial resynthesis of PI, PIP, and PIP2. Inhibition of these lipid phosphatases by lithium has led to the hypothesis that lithium therapy may deplete cellular inositol (i.e., myoinositol), thus interfering with IP3-mediated cell signaling (15).
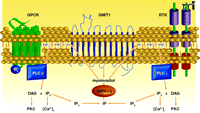
The phosphatydilinositol cycle in signal transduction. G protein–coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs) mediate the activation of phospholipases C–(PLC)-β and -γ), which cleave phosphoinositide bis- phosphosphate (PIP2) to yield diacylglycerol (DAG) and inositol trisphosphate (IP3). DAG and IP3 activate protein kinase C (PKC) and release calcium [Ca2+ ]i from intracellular stores, respectively. IP3 is rapidly hydrolyzed to inositol bisphosphate (IP2) and inositol monophosphate (IP). Inositol monophosphatase-(IMAP)-1 and -2 hydrolyze IP to regenerate myoinositol. Inositol gains access to the cellular cytoplasm via uptake by the sodium/inositol cotransporter (SMIT1).
Two isoforms of inositol monophosphatase—IMPA1 and IMPA2—have received considerable attention. Both IMPAs function as homodimers and require magnesium for enzymatic activity (20, 21), and it has been postulated that lithium may inhibit IMPAs by competing with magnesium for binding at the active site (22). Alternatively there are also lines of evidence that displacement of magnesium by lithium may prevent the dimerization of IMPAs (32).
Lithium and Inositol Homeostasis: In Vivo Evidence
In Xenopus, lithium interferes with embryonic development, resulting in the duplication of dorsal and anterior structures; in Dictyostelium lithium interferes with the life cycle of this slime mold and leads to an expanded stalk cell population. In both organisms, lithium induces inositol depletion, and its metabolic effects are antagonized by addition of exogenous inositol (15, 19). Furthermore, lithium—albeit at concentrations three- to fivefold above therapeutic doses—and other mood stabilizers have been reported to induce inositol depletion and trigger growth cone collapse in primary neuronal cultures (24). Finally, chronic lithium treatment in rodents reproducibly reduces the inositol content of various brain tissues by about twenty to twenty-five percent (25, 26). Although these observations point toward a possible involvement of inositol depletion in the effects elicited by lithium, attempts to replicate the behavioral effects of lithium in mammals by inducing brain inositol depletion have been less conclusive.
Lithium and Inositol Depletion: Behavioral Studies
Mutational inactivation of the IMPA1-encoding gene (Impa1) in mice results in an embryonic lethal phenotype that can be rescued by administration of inositol to the mother during pregnancy and lactation (27). In contrast, Impa2-knockout mice develop normally (28). Curiously, deficiency in either Impa1 or Impa2 does not significantly affect brain inositol content in adult mice (27, 28). When submitted to a battery of tests to evaluate depressive-like (e.g., tail-suspension and forced-swim tests) and manic-like behaviors (e.g., amphetamine-induced locomotor activity), Impa2-knockout mice show no significant behavioral differences relative to their wild-type littermates (28), thus failing to replicate typical antidepressant- and antimanic-like effects of lithium ( 18, 29, 30). In contrast, inositol-rescued Impa1-knockout mice display locomotor hyperactivity, a behavior that is normally antagonized by lithium in rodents (1, 27, 30). Unfortunately, the hyper-active phenotype of the Impa1-knockout mice precludes their utility in addressing antide-pressant agents, as changes in activity can significantly confound such studies (27). Finally, it is of interest that Impa1-knockout mice, like lithium-treated wildtype mice, display enhanced sensitivity to pilocarpine, a seizure-inducing agent (27).
Apart from recycling of phosphoinositol, cellular uptake by the sodium/inositol cotransporter (Smit1) is a major source of inositol in cells. Smit1-knockout mice die during embryogenesis but can be rescued into adulthood by inositol supplementation during pregnancy (31). Further investigations conducted in Smit1 hemizygous mice show that Smit1 gene expression correlates to levels of brain inositol; brain inositol levels in hemizygous mice (60–70% of the wild-type level) are slightly superior to those obtained following chronic lithium treatment (75–80%)(25). Despite this similarity of effect on inositol brain levels, Smit1 hemizygotes do not display behavioral responses similar to those of lithium-treated mice in tests of depressive-like behaviors (25). Furthermore, extensive depletion of inositol (> 90%) in the brains of Smit1-knockout mice elicit no dramatic effects on phosphatidylinositol (PtdIns) levels, thus indicating that the effect of inositol depletion on PIP2- and IP3-mediated signaling in vivo may be less important than previously thought (32, 33). Finally, it is of interest that rescued Smit1-knockout mice have enhanced sensitivity to pilocarpine, thereby replicating a phenotype of Impa1-knockout mice (34).
Lithium and the Role of Inositol Depletion: Summary
The inositol depletion model of lithium action represents one of the original attempts to explain, in molecular terms, its therapeutic action. Despite its common appeal, the relevance of the model has been questioned by in vivo studies. Inositol depletion that is more drastic than that resulting from treatment with lithium fails to affect predicted behaviors (25). Interestingly, studies conducted in either Impa1- or Smit1-knockout mice suggest that inositol depletion contributes to the enhanced sensitivity to pilocarpine that is induced by lithium. Still, the relationship between pilocarpine sensitization and effects on mood is not clear, and indeed, other mood stabilizers, such as valproic acid, have an anticonvulsant effect in the pilocarpine paradigm (35). Despite these strong reservations, one must consider that perturbations of inositol homeostasis, such as those evoked by lithium, may exert greater behavioral effects in humans with bipolar disorder than they do in mice. Furthermore, it is possible that even a small depletion of inositol, within the context of other molecular targets in the bipolar brain, may still be crucial to the effects of lithium on behavior.
Inhibition of Glycogen Synthase Kinase 3 (GSK3)
A second compelling mechanism for the action of lithium was suggested by the discovery in the mid 1990s that lithium is also an uncompetitive inhibitor of members of the GSK3 family (16, 17). The therapeutic relevance of these findings, however, has remained unclear, given the high Ki (~3.5mM for GSK3α and ~2.0mM for GSK3β) of lithium for GSK3 isoforms (19, 36). GSK3α and GSK3β are closely related serine/threonine kinases originally implicated in the regulation of glycogen synthesis in response to insulin (37, 38). Each is constitutively active and can be inactivated through the phosphorylation of a single serine residue within the N-terminal regulatory domain (Ser 21 of GSK3α; Ser9 of GSK3β) (38). The serine/threonine kinase Akt, also termed protein kinase B (PKB), has been shown to inhibit GSK3α and GSK3β (Figure 2⇓) in response to multiple hormones and growth factors, including BDNF, IGF, and insulin (38–40). Akt is regulated through phosphatidylinositol-mediated signaling. Figure 2⇓ shows the activation of Akt according to two steps: 1) its recruitment to the plasma membrane by phosphorylated phosphatidylinositol [Ptdlns(3,4,5) P3, Ptdlns(3,4)P2]; and 2) its subsequent phosphorylation at Thr308 (catalyzed by PDK1) and Ser473 (catalyzed by PDK2) (41–44). GSK3 isoforms have been implicated in multiple physiological functions, including glycogenesis, embryonic development, Wnt signaling, and apoptosis, as well as dopaminergic and serotoninergic neurotransmission (30, 38, 45, 46).
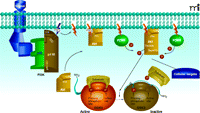
The extracellular signal–dependent Akt regulation of glycogen synthase kinase function. Extracellular signals through RTK and GPCR activate phosphoinositol-3 kinase (PI3K), which converts PIP2 to PIP3 and directs Akt to the plasma membrane via interaction of its PH domain with PIP3. Akt is then activated by phosphorylation by phosphatidylinositol dependent kinase-(PDK)-1 and -2. Akt can then phosphorylate a series of cellular substrates. See text for details.
The mechanism by which lithium inhibits GSK3 activity is still incompletely understood. One attractive theory is that lithium acts as a competitive inhibitor for the binding of the cofactor magnesium (36). Competition for the binding of magnesium has also been suggested as a mechanism for the lithium-mediated inhibition of IMPAs (see above). Indeed, magnesium and lithium ions share similar ionic radii (0.065 and 0.060 nm, respectively) (47). The proposed competitive mechanism of inhibition by lithium predicts that the in vivo inhibition of GSK3 may be greater than inhibition levels observed in in vitro studies that generally monitor GSK3 activity at optimal concentrations of magnesium ion (36).
In Vivo Evidence of GSK3 Inhibition by Lithium
Multiple independent lines of evidence indicate that both acute and chronic lithium treatment can reduce GSK3 activity in vivo. Early studies conducted in cell culture systems have shown that lithium prevents the phosphorylation of proteins that are substrates for GSK3, such as the microtubule-associated proteins tau and MAP-1B as well as β-catenin, a transcription factor in the Wnt pathway (16, 17, 19, 48). Furthermore, inhibition of GSK3 in Xenopus and Dictyostelium recapitulates the effects of lithium (see above) (17). More recently, multiple studies of neurodegenerative and neuroinflammatory processes have shown that chronic lithium treatment produces effects that are compatible with the inhibition of GSK3 as observed in the mouse central nervous system (9–11, 49). Finally, lithium treatment increases the expression of β-catenin in the mouse brain (18, 50, 51). Given that the degradation of cytoplasmic β-catenin is promoted upon its phosphorylation by GSK3 (52), increased β-catenin levels in response to lithium have generally been attributed to reduced GSK3 activity, but a direct role for GSK3 inhibition in therapeutic responses to lithium has not been established. Apart from its action as a GSK3 inhibitor, lithium has also been reported to activate neuronal Akt, a regulatory kinase that phosphorylates and thereby inhibits GSK3 (30, 53–55). In this way, it is conceivable that reduced GSK3 activity, as a component of lithium therapy, could reflect the targeting of either or both kinases, depending on dosage.
Evidence for GSK3 Inhibition in Lithium-Regulated Behavior
Both direct and circumstantial lines of evidence support an involvement of GSK3 in the regulation of symptoms of mania and depression by lithium. Research conducted in our laboratories has shown that GSK3 is involved in the regulation of behavior by the monoamine neurotransmitters dopamine and serotonin (5-HT), which are important pharmacological targets for the management of depression and psychosis (18, 30, 46, 56–69). Furthermore, various kinase inhibitors, acting directly on GSK3 or indirectly to limit its activity, replicate the effect of lithium in in vivo models of depression and mania (18, 30, 46, 60). Finally, genetic down-regulation of GSK3 in GSK3β haploinsuficient mice recapitulates many behavioral effects of lithium (30, 51), whereas overexpression of a Ser9 mutant of GSK3β, which results in constitutive GSK3 activity, reproduces behavioral correlates of hyperactivity and mania (61).
Lithium and the Role of GSK3 Inhibition: Summary
Recent literature provides strong evidence for the involvement of GSK3 inhibition in the regulation of behavior by lithium, and there is no doubt that under certain conditions lithium inhibits GSK3. Nevetheless, the relative degrees to which lithium functions as a direct inhibitor of GSK3, as opposed to acting as a regulator of Akt activity, are not clear in a behavioral context. One way out of this conundrum would be to conduct behavioral studies on transgenic mice that lack the N-terminal regulatory serine residues of GSK3α and β (62). Such mice would express GSK3 isoforms that cannot be inhibited by Akt and should thus constitute a good model to sort out the contribution of direct GSK3 inhibition in the behavioral effects of lithium. Another point of caution is that, because mice that lack a functional Gsk3b allele die during development (63), most behavioral studies of lithium that support the involvement of GSK3 have been conducted in GSK3β haploinsuficient mice. These mice are useful for studying the contribution of GSK3 to some behaviors, but it should be noted that they express normal levels of GSK3α and show only about 25% reduction in total GSK3 activity (30). Therefore, the effects of lithium observed in GSK3β haploinsuficient mice may be significantly influenced by variations in experimental conditions, whereas therapeutic effects of lithium may be predicated on the existence of two GSK3 (30, 46, 59). There are presently no animal models in which the behavioral effects of lithium can be assessed in the total absence of neuronal GSK3. Obviously, animal models in which both GSK3 isoforms could be engineered and experimentally manipulated would be greatly help to elucidate the mechanism of lithium action with respect to GSK3 activity.
A Network of Effects: the Akt·βArr2·PP2A Protein Complex as a Lithium Target
The initial evidence for lithium as an upstream regulator of GSK3 came from cellular studies examining the ability of lithium to protect against neuronal glutamate toxicity (54) and from in vivo paradigms examining signaling responses to mood stabilizers in rodents (30, 53). Direct biochemical evidence has further established that lithium can regulate Akt and GSK3 in vivo by disrupting the formation of a protein complex involved in GPCR signaling (18, 30, 58, 64).
Following stimulation and activation of their cognate G proteins, GPCRs are rapidly phosphorylated by members of a family of GPCR kinases (GRKs), leading to uncoupling of G proteins and the recruitment of β-arrestins (Figure 3⇓) (65–67). The interaction of β-arrestins with GPCRs, which limits the ability of the GPCR to signal further through the G protein, is followed by the formation of an endocytic complex that promotes the internalization of receptors via clathrin-coated pits (65, 67–69). However, the role of β-arrestins in GPCR regulation is not limited exclusively to desensitization. It has become apparent that apart from their canonical functionality through G proteins, GPCRs can also elicit cellular responses mediated by the formation of signaling protein complexes scaffolded by β-arrestins (70–72).
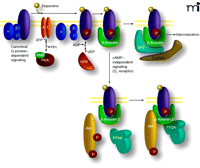
Dual role of β–arrestins in mediating GPCR desensitization and G protein–independent signaling. Activation of dopamine receptors (DAR) activates G proteins, which leads to activation (Gαs) or inhibition (Gαi/o) of adenylyl cyclase and modulation of the cAMP-dependent protein kinase PKA (G protein–dependent signaling). This is rapidly followed by receptor phosphorylation by GRKs and the recruitment of β–arrestins, leading to termination of G protein signaling and formation of an internalization complex formed by β–arrestin 1 and/or β–arrestin 2, AP2, clathrin. Recruitment of β–arrestin 2 following activation of D2-class receptors also results in the formation of a signaling complex composed at least of β–arrestin 2, PP2A, and Akt, which results in a deactivation of Akt by PP2A and subsequent stimulation of GSK3-mediated signaling.
The D2-like dopamine receptors (including the D2, D3, and D4 dopamine receptors) are a small family of GPCRs that couple to the Gαi/o protein, whereby receptor activation leads to a reduction in the production of the second messenger cyclic adenosine monophosphate (cAMP) (73–74). Prolonged stimulation of D2 receptors in the mouse striatum causes the dephosphorylation and concomitant inhibition of Akt, which in turn results in the activation of both GSK3 isoforms (Figure 4⇓). Behavioral and biochemical evidence has revealed that regulation of Akt/GSK3 signaling by the D2 receptor is not affected by cAMP, but is instead mediated by the formation of a protein complex that includes Akt, βArr2, and the heterotrimeric protein phosphatase 2A (PP2A), which facilitates the dephosphorylation and concomitant deactivation of Akt by PP2A in response to dopamine (Figures 4⇓ and 5⇓) (30, 64, 77).
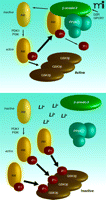
The effect of lithium on interactions among Akt, βArr2, and PP2A. Under basal conditions (upper panel) phosphorylated/activated Akt is efficiently dephosphorylated, leaving GSK3 mostly in an unphosphorylated/ active state. During acute or chronic lithium treatment (lower panel), lithium destabilizes the Akt·βArr2·PP2A complex, augmenting the steady state of active Akt and promoting inactivation/phosphorylation of GSK3 in neurons.
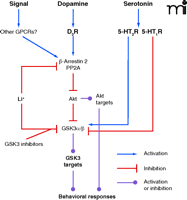
Modulation of the Akt and GSK3 signaling pathway by various GPCRs. Signals from dopamine D2 receptors, serotonin receptors, and other GPCRs may modulate this pathway either through a β-arrestin-dependent or -independent mechanism. Lithium inhibits GSK3 either by enhancing Akt phosphorylation by interfering with the D2 receptor–dependent Akt·βArr2·PP2A complex or by acting as a direct inhibitor of GSK3.
Lithium and the Akt·βArr2·PP2A Complex
In contrast to most previously described approaches to infer the molecular mechanism of lithium action, data supporting a role of the Akt·βArr2·PP2A complex were obtained primarily from in vivo models used to study the effect of mood-altering drugs. Acute lithium treatment in mice antagonizes the development of dopamine-dependent locomotor behaviors by interfering with regulation of the Akt/GSK3 signaling through D2 receptors (29, 30, 78). This interference can be replicated by preventing the formation of the Akt·βArr2·PP2A protein complex. In βArr2-knockout mice, the absence of the Akt·βArr2·PP2A abolishes regulation of Akt/GSK3 signaling by D2 receptors and an increases basal phosphorylation of Akt (Thr308) and GSK3β (Ser9), similar to observations following lithium treatment (18, 30, 64). Furthermore, when administered acutely or chronically to βArr2-knockout mice, lithium fails to activate Akt and thus cannot inhibit GSK3 as it does in the striatum of wild-type animals (18). Accordingly, striatal β-catenin levels in the βArr2-knockout mice fail to rise in response to chronic lithium treatment (18). Significantly, all the βArr2-knockout characteristics described here correlate with the biochemical effects of lithium upon the formation of the Akt·βArr2·PP2A signaling complex. A series of co-immunoprecipitation experiments have shown that therapeutically relevant lithium concentrations (0.5 to 1mM) are sufficient to destabilize the Akt·βArr2·PP2A complex in vitro as well as in the brains of living mice (18). Remarkably, lithium appears to be selective in destabilizing the Akt·βArr2·PP2A complex, as other protein complexes scaffolded by βArr2 and involved in GPCR desensitization or signaling are unaffected by lithium (18).
The exact mechanism through which lithium interferes with the formation of the Akt·βArr2·PP2A complex awaits further investigation; however, preliminary in vitro experiments with recombinant purified Akt1 and βArr2 suggest that interactions of these two proteins required for complex formation is magnesium-dependent. Thus, competition between lithium and magnesium for binding to at least one of the components could underlie the instability of the complex in the presence of lithium (18, 47).
Lithium and the Akt·βArr2·PP2A Complex in Behavior
As discussed above, several behavioral studies implicate GSK3 inhibition in the effects of lithium (30, 51, 59–61). In addition, behavioral responses in βArr2-knockout mice indicate that regulation of GSK3 by the Akt·βArr2·PP2A protein complex is key. When evaluated for dopamine- or psychostimulant-induced locomotor activity, βArr2-knockout mice are reminiscent of lithium-treated wild-type mice, displaying generally reduced responses (18, 30, 64). Furthermore, neither acute nor chronic lithium treatment has significant effects on the behaviors of βArr2-knockout mice (18). Intriguingly, despite their resistance to lithium, βArr2-knockout mice remain behaviorally responsive to a selective and direct inhibitor of GSK3 activity, thereby suggesting that the Akt·βArr2·PP2A protein complex is an upstream modulator of GSK3 in the signaling cascade (See Figures 3⇑ and 4⇑) that regulates lithium-sensitive behavioral responses (18).
Lithium and the Akt·βArr2·PP2A Protein Complex: Summary
The Akt·βArr2·PP2A protein complex appears to satisfy many of the prerequisites of a direct lithium target involved in the regulation of behavior. It is directly affected by lithium both in vitro and in the brain, and its elimination precludes both biochemical and behavioral effects of lithium. It is plausible that additional mechanisms contribute to the pharmacological effects of lithium on behavior. Furthermore, the relevance of lithium-mediated Akt·βArr2·PP2A complex disruption in the effects of lithium therapy remains to be established. A related question pertains to the selectivity of lithium for the Akt·βArr2·PP2A complex. Lithium does not affect general functions of βArr2 in GPCR desensitization and signaling; however, βArr2 has multiple interaction partners (79), and the possibility that lithium will undermine the interaction of other proteins with βArr2 must be considered. A second question relates to the regulation of the Akt·βArr2·PP2A complex by dopamine D2 receptors; specifically, it is possible that other GPCRs may also signal through the Akt·βArr2·PP2A protein complex. Indeed, depression-like behaviors that are affected by lithium have generally been linked to 5-HT- and norepinephrine-based neurotransmission (80). Finally, regulation of Akt activity by lithium raises the possibility that substrates other than GSK3 may also play a role in lithium action.
GSK3 Substrates and Behavioral Regulation
The data discussed above establish that at least two potential mechanisms of lithium action converge on the modulation of the Akt/GSK3 signaling pathway. Although these enzymes are integration hubs for many signaling modalities, the identity of the downstream targets that may regulate behavior has remained elusive. Akt and GSK3 both have multiple substrates, including proteins involved in metabolism, cell survival and death, cytoskeletal organization, and gene regulation (38, 43, 81, 82). Below we provide a brief overview of possible targets that have been investigated in the context of mood-stabilizing drug effects.
β-Catenin
β-catenin is a multifunctional protein that acts both as a transcription factor and a scaffolding molecule. In this latter function, β-catenin interacts with cadherins and α-catenin and thereby regulates cytoskeletal organization of adherens junctions. Formation of such complexes may play a role in synaptic plasticity, because β-catenin is recruited to dendritic spines following depolarization (83). Moreover, a reduction in cytoplasmic levels of β-catenin and other members of the catenin·cadherin complex can reduce dendritic arborization in cultured hippocampal neurons (84). In the Wnt signaling cascade, phosphorylation of monomeric β-catenin by GSK3 leads to its ubiquitination and proteosomal degradation (52). In this way, the modulation of GSK3 by monoamines and psychotropic drugs may lead to changes in β-catenin levels that affect synapse morphology and gene expression. Interestingly, chronic lithium treatment raises β-catenin expression in different regions of the mouse brain (18, 50, 51). Furthermore, overexpression of β-catenin in transgenic mice recapitulates the effects of lithium hyperactivity and depression models in several relevant behavioral paradigms (50). However, the tissue-specific knockout of β-catenin in the adult mouse forebrain has little behavioral consequence (85) These studies collectively suggest that β-catenin may be important for the action of psychotropic drugs, whereas it may play a lesser role in regulation of normal behavior by mono-amine neurotransmitters.
Glutamate Receptors
Akt/GSK3 signaling appears to regulate synaptic plasticity and ionotropic glutamate receptor functions. Importantly, these receptors, and glutamate neurotransmission in general, have been strongly implicated in the presentation of psychiatric disorders such as schizophrenia (86–89). Mood stabilizers have also been reported to affect glutamate neurotransmission (90). The expression and trafficking of the heteromultimeric ionotropic glutamate receptors are closely regulated by complex networks of signaling molecules and scaffolding proteins (91). Dopamine and 5-HT may also affect synaptic plasticity by regulating the expression, phosphorylation, and trafficking of ionotropic glutamate receptors and associated proteins in neurons (92–94). In addition, to the extent that its activation level has been correlated to long-term potentiation and long-term depression, GSK3 has been linked to glutatmatergic activity (95–97) and affects the trafficking of NMDA receptor subunits to the cell surface (95, 98).
Circadian Rhythm Proteins
The regulation of the fly GSK3 ortholog shaggy by 5-HT modulates circadian entrainment (99). Intriguingly, disruption of circadian rhythm has been proposed to play a role in diverse mental illnesses (100, 101). Exposure of cells to lithium appears to interfere with transcription of the clock gene Bmal1. Dopamine D 2 receptors, which regulate the activity of GSK3 (see above), also affect circadian rhythm regulated gene expression and behavior (102–104), and GSK3β has been shown to regulate mammalian circadian protein functions in cultured cells (105).
Arachidonic Acid
Chronic lithium treatment in rats has been shown to reduce brain arachidonic acid turnover by a mechanism involving downregulation of cycloxygenase 2 (COX-2) (106, 107), an effect also attributed to chronic treatment with the mood stabilizers valproic acid and carmabazepine (108, 109). [Indeed, a recent clinical trial indicates that the COX-2 inhibitor clecoxib can potentiate the effects of antipsychotics and mood stabilizers in individuals with bipolar disorders (110).] Carmabazepine appears to affect arachidonic acid signaling by interfering with dopamine D2 receptor functions (111), and COX-2 expression is sensitive to GSK3 activity in some cell culture systems (112) and mouse peripheral tissues (113–114).
Conclusion
Since the realization, nearly sixty years ago, that lithium can exert mood-stabilizing effects, different molecular mechanisms have been proposed to explain its actions. Here, we have reviewed three of these putative mechanisms. Compelling lines of evidence suggest that lithium can reduce brain inositol, inhibit brain GSK3 activity, and disrupt the formation of the Akt·βArr2·PP2A in dopaminoceptive brain neurons. For all three putative mechanisms, lithium may act by competing with magnesium binding. Lithium has also been suggested to affect the functions of G proteins in a magnesium-dependent way (115). However, mechanisms governing the specificity of lithium/magnesium competition are still poorly understood (116), and the relevance of lithium/magnesium competition to the therapeutic effects of lithium awaits elucidation. Furthermore, the possible identification of other proteins or protein complexes affected by this mechanism in the regulation of behavior represents an interesting avenue for future research.
Acknowledgments
This work was supported in part by a Canada Research Chair in Molecular Psychiatry and a Human Frontier Science Program Career Development Award (to J-MB) and National Institutes of Health Grants DA-13511, NS-19576, MH-73853 and MH-40159 (to MGC). Unrestricted gifts from the Lennon Family Foundation and Lundbeck Research USA to Duke University are greatly appreciated.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Marc G. Caron, PhD, is James B. Duke Professor of Cell Biology and holds secondary appointments in Medicine and Neurobiology at Duke University Medical Center. His long-standing research interests have concerned G protein–coupled receptors and neurotransmitter transporters. His recent efforts have used genetic approaches to develop animal models of neurobiological disorders.

Jean-Martin Beaulieu, PhD, is Assistant Professor and Canada Research Chair in Molecular Psychiatry at Laval University/ CRULRG in Québec City. His research on dopamine receptor signaling in vivo has led to the identification of mechanisms of action for lithium and antipsychotics. He continues to research how changes in signaling processes participate in the regulation of behavior by various psychiatric drugs.

